Information Technology Department College of Computer and Information Sciences
King Saud University Riyadh, Saudi Arabia.
Corresponding author email: halshamlan@ksu.edu.sa
Article Publishing History
Received: 15/02/2023
Accepted After Revision: 18/03/2023
Cardiovascular disease (CVD) is the world’s premier cause of morbidity and death. CVD is a class of heart or blood vessel diseases. CVD contains the coronary artery disease (CAD) such as unstable angina (UA) and myocardial infarction (MI) diseases. Clinicians use additional tools to support clinical evaluation and improve their ability to detect the susceptible patient at threat for CVD. Biomarkers are one such method to identify potential risk persons, rapidly and reliably diagnose disease symptoms that efficiently predict and treat disease. Discovery of MicroRNAs (miRNAs) representing a class of small, non-coding RNA molecules opens interesting opportunities to use the patterns of miRNAs as a biomarker for cardiovascular diseases. The objective of this study is to define miRNA and genes potentially associated with MI.
Rothman dataset includes 52 samples of Acute Coronary Syndromes (ACS). including 18 patients with myocardial infarction (MI) and 8 patients with unstable angina (UA). Overall (number of genes selected) candidate ncRNA biomarkers have been defined and a ncRNA-based classifier has been created to predict MI risk which based on 7 ncRNA expression data using vector support machines SVM and decision tree classifiers. The experimental results obtained through applying these mechanisms on the Rothman dataset. The classification model’s performance is evaluated using the V-fold validation and LOOCV methods. The outcome of this search can be used by the drug designer for pathway analysis and CVD treatment decisions.
Cardiovascular disease, Myocardial infarction, Biomarkers, Gene expression, Classification.
AlShamlan. H. M. Using Gene Expression Profile to Extract the Biomarker Genes of Cardiovascular Disease. Biosc.Biotech.Res.Comm. 2023;16(1).
AlShamlan. H. M. Using Gene Expression Profile to Extract the Biomarker Genes of Cardiovascular Disease. Biosc.Biotech.Res.Comm. 2023;16(1). Available from: <a href=”https://bit.ly/3JPYuhb“>https://bit.ly/3JPYuhb</a>
INTRODUCTION
Cardiovascular disease is one of the world’s leading causes of death, including coronary artery disease which resulting due to reduced blood flow into the coronary arteries caused by thrombus formation. The consequences of coronary vasospasm, which may lead to myocardial infarction (MI) as a result of myocardial tissue necrosis. Many CVD patients are not diagnosed immediately, and the prognosis is very poor, as stated in 2013, the total CVD death rate was 222.9 per 100,000 individuals (Mozaffarian et al., 2016). Several researches have been conducted in the field of genetic biomarkers for CVD, and circular RNAs have obtained significant attention due to their clinical and biological use in the diagnosis and treatment of CVD.
Over the past few decades, patients have usually been tested with an initial assessment and assessed with a risk rating or prediction algorithm that takes into account medical history, physical examination and other indicators (Čulić et al., 2002). Additional tests, including electrocardiogram (Slater et al., 1987), coronary computed tomographic angiography (Goldstein et al., 2011) were applied to these evaluations. The existing methods, however, are inadequate for a precise diagnosis and highly sensitive, particularly in the distinction between MI and UA. Furthermore, there is a silent myocardial infarction phenomenon that is predicted to happen in about 64% of cases where patients have no chest pain or any other symptoms (Valensi et al., 2011, Gangwar 2017, Zaiou & Bakillah 2018, Guo et al. 2019 and Xu & Yang 2021).
Recent years have seen a growing interest in studying the mechanism of gene expression in which DNA instructions are translated into non-coding RNAs (ncRNAs), that are regarded to be essential epigenetic regulators of several physiological and pathological conditions, including cardiovascular diseases such as the acute coronary syndrome. Massive attempts have been made to use these RNA molecules as predictive biomarkers for a number of diseases including cardiovascular disease (Gangwar 2017, Zaiou, Bakillah 2018 Guo et al., 2019, Xu & Yang 2021).
MATERIAL AND METHODS
Microarray technique was used to measure the expression of genes. The method of selection of features that led to a reduction in the volume of data used to identify biomarkers which improves the performance of the diagnostic workbook. So far, very few studies have looked at ACS subtype MI and UA, in terms of mRNA expression as a biomarker of Cardiovascular disease. In this project, I have used MI and UA samples that are sorted based on specific features. Then I filtered these samples by using information gain algorithm to select features. The central objective of this research is to classify gene expression using classification algorithm to extract the biomarker of CVD. In this research, I have applied filter and wrapper methods to the analysis of the microarray gene expression profile. In addition, there is a previous study that used the same dataset but different classification methods to distinguish the biomarkers of cardiovascular disease. The aim of this study is to improve the accuracy of the previous study by using different classification methods, which it exceeded by the percentage of accuracy 100 % with SVM and 91% with decision tree.
Literature Review: In order to present the various algorithms that have been examined. Finding the impacted and overexpressed genes for the diseases and then using association rules and gene intervals to classify gene expression. For this purpose, the researcher must know the basic knowledge for the implementation of bioinformatics strategies, including statistical methods, genetic selection techniques, associative classification algorithms and cross validation methods. Kazmi & Gaunt (2016) discussed the genomic tools and technologies that used to extract the biomarker of heart disease by analyzing the gene expression of a blood cells. The initial features were discovered by fitting a linear model for each probe collection across all arrays of patients with myocardial infarction and healthy individuals.
Three separate feature optimization algorithms were developed that specified two different sets of genes, one using MI and unstable angina, and the other using MI and normal controls. The experimental results show that the analysis of gene expression profile with the microarray study is successfully diagnoses biomarkers of some heart disease including myocardial infarction disease. It has also been shown to be effective in discriminating myocardial infarction in patients with clinical symptoms of cardiac ischemia but without stable coronary artery disease or myocardial necrosis, (Guo et al. 2019 and Xu & Yang 2021).
The limitation in the study was that the specificity measures for some experiments could not be fully justified due to a limited number of samples. In (Cheng et al., 2017) the author presented the reasons for coronary artery disease (CAD) leading to increased mortality, angina and myocardial infarction. The dataset was processed by identifying the differential expressed genes (DEGs) and then clustering the result using the pheatmap package in R. They show the protein coding gene MAP1B and the ARG1 gene that provides instructions for the production of the enzyme arginase that appears to be indirectly regulated by CDKN2B-AS through miR-92a in the pathogenesis of CAD. This research needs to be improved through the use of empirical research to confirm these results. Wu et al., (2018) reported on the causes and treatment of myocardial infarction (MI) by analyzing miRNA that represent a signature expression pattern of miRNA that plays a vital role in MI, and also by analyzing the protein-protein interaction network, which plays a role in the understanding of the MI molecular mechanism.
The results of the experiment have been validated using RT-PCR. The relationship of the non-coding miRNA to the expression of target genes. This study contributes to the understanding of the MI mechanism by supporting clinical diagnostic and the development of appropriate treatment for MI. The drawbacks of this study in the lack of research samples and the need for confirmation of this analysis by experts from other disciplines such as cytology. Lu et al., (2018) reported that there’s little understanding about the patterns and function of lncRNA in the interduce of acute coronary syndromes (ACSs). Dysregulated lncRNA expression has been involved in cardiovascular disease. The lncRNA expression profiles were examined in the two distinct clinical entities of ACS, unstable angina (UA) and myocardial infarction (MI). In addition, functional analysis mentioned that these candidate lncRNA biomarkers could be implicated in known MI-associated pathways and biological processes. This research was the first in ACS growth to recognize altered patterns of lncRNA expression. this paper presents a novel functional lncRNAs may be used as therapeutic targets biomarkers and candidate diagnostic.
This study can be improved by conducting more experimental studies to determine the molecular mechanism of these novel lncRNAs in the progression of ACS. In (Guo et al., 2019) this study, gene data for patients with UA or MI have been used to define optimal pathways that provide extensive information for the progression of MI/ UA. The paper mentions the limitations of the existing method in disease progression information obtained from the identification of differentially expressed genes (DEGs). The paper’s contribution was made by using the LIMMA Detection Package (DEGs) between UA and MI, in addition to the differential co-expression network (DCN) and the sub-DCN for the DEGs. (DCN) proved to be a new holistic approach to microarray analysis. The importance of identifying the optimal pathways to demonstrate the progress of ACS development. This work needs to discover the underlying mechanisms for the progression of ACS using animal models.
Table 1. The datasets selection and classification methods that used in related works.
| Accuracy | genes | Classification | Feature Selection | Dataset | Related Works | |||||||||||||||||||||
| GSE62646 | GSE49925 | GSE48060 | GSE29532 | GSE29111 | GSE20680 | GSE20681 | ||||||||||||||||||||
| 100%
88% |
7
9 |
-k-nearest neighbor (KNN)
-(LOOCV) |
P-Value
t-tests |
* | * | * | * | (Kazmi, N., et al. 2016) | ||||||||||||||||||
| 84.6%
90.8% 96.9% |
52
87 41
|
-(SVM) | p-value
-Recursive feature elimination (RFE) algorithm |
* | * | (Cheng, M., et al. 2017) | ||||||||||||||||||||
| 95%
70% 94% |
48
21 10
|
generalized linear modeling (GLM)
|
P- value
– Gene Ontology (GO) and Kyoto -Encyclopedia of Genes Genomes (KEGG)
|
* | * | (Wu, K., et al. 2018) | ||||||||||||||||||||
| 90.38% | 7 | -Random forest
– SVM -(LOOCV) (ROC) (AUC) |
-DAVID Bioinformatics Tool
-(GO) -Kyoto Encyclopedia of Genes and Genomes (KEGG) |
* | (Lu, Y., et al. 2018) | |||||||||||||||||||||
| 80% | 3 | – (SVM)
-GBA pathway prediction method -the mean AUC across all pathway |
– DAVID tool
– fold-changes (FC) with log2 base (logFC) |
* | (Guo, S. Z., et al. 2019) | |||||||||||||||||||||
Table 2. Microarray Datasets details used on related works.
| Dataset | URL | Samples | Genes | Database |
| GSE20681 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE20680
|
198 | 45015 | NCBI Gene Expression Omnibus (GEO) |
| GSE20680 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE20680
|
195 | 45015 | |
| Rothman Dataset: GSE29111 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE29111 | 52 | 54675 | |
| Gregg Dataset: GSE49925 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE49925 | 338 | 14111 | |
| Beata Dataset: GSE62646 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62646 | 98 | 33297 | |
| Nelson Dataset: GSE48060 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48060 | 52 | 54675 | |
| GSE29532 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE29532 | 55 | 22011 |
Proposed Method: Bioinformatics is the process of collection, classification, storage, and analysis of biochemical and biological information using computers especially as applied to molecular genetics and genomics. In this section, we will define the methods that we applied on dataset which are a feature selection method for biomarker mining and classification methods which is the concept of statistical models for the diagnosis of diseases. These methods calculate the mutual information between the candidate and the class label and the average of the mutual information between the candidate and the attributes of the selected subset.
In this research paper, we have used Information Gain (IG) feature selection method and with Decision tree classification method for selecting important genes to reduce the number of genes that help to increase the speed and accuracy of prediction systems. Then we compared the resulting accuracy with SVM classification methods using LOOCV cross validation. Below we will illustrate brief description about each method. Figure 1, shows clear view about our proposed method.
Figure 1: Proposed Algorithm
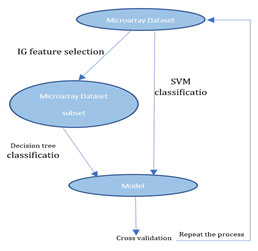
Feature Selection Method: For feature selection methods we used both the filter model and the wrapper model. Information gain filter feature selection method and wrapper decision tree selection method. Filter methods use heuristics based on data characteristics to select predictive subsets of features. The wrapper model uses a mining algorithm’s predictive accuracy to evaluate a selected subset’s goodness.
a) Information gain (IG): Information gain is applied as a filter in the selection of genes. It is measured by calculating the IG value by using a system entropy. Then the genes are ranked depend on its IG value. The information gain calculates the difference between the entropy before splitting and the average entropy after splitting the samples based on the given attribute values. Entropy is the proportion of the instances to its classes that calculate as the following equation: IG = information before splitting (parent) — information after splitting (children)
Classification Methods: In this research we applied two classification methods. The first one is support vector machine (SVM) which is directly applied to microarray dataset. The second one is decision tree (DT) algorithm which is applied as wrapper method combined with information gain (IG) method.
a) Support Vector Machine (SVM): Support vector machine (SVM) is a category of associated supervised learning methods used in regression and classification. The simplest method (SVM) is a linear classification, which is intended to draw a line that separate data into two-dimensional.
b) Decision Tree (DT) : The decision tree is hierarchically represented with the root at the top and split into branches / edges until the end of the branch that no longer splits is the decision / leaf. The structure of decision tree where the branches represent conjunctions and the leaves represent classifications of features. Previous assumptions about the nature of the data are not required, therefore this method can classify the numerical and categorical data. The output attribute must be unique in the decision tree algorithm. In the situation of changing training data, different attributes selection produced with selecting point in the tree.
Cross validation: It is necessary to test data samples independently of the learning dataset used to build a classifier, in order to perform a classification error measurement. V-fold cross-validation technique performs independent tests without requiring a separate test dataset that lead to increases the difficulty and price of the test and use this technique without reducing the data was using to construct the tree. The learning database is split into 10 folds of cross-classification rows groups.
a)Leave-one-out cross-validation (LOOCV): Includes the sample divided into validation data for a single observation and training data for the remaining observations. This contributed to the quality of the classifier based on lncRNA, including its precision, sensitivity and specificity.
RESULTS AND DISCUSSION
In this section we will discuss the gene expression dataset used in our research. Then, we will present the experimental result of our proposed method and comparison with more related work. In this research, (GEO; GSE29111) Rothman dataset includes 52 samples of Acute Coronary Syndromes (ACS). including 18 patients with myocardial infarction (MI) and 8 patients with unstable angina (UA). The number of (MI) samples is 26 and the number of (UA) samples is 16. The total number of the attributes are 54675.
The Rothman dataset contains samples of whole blood mRNA expression obtained from 26 acute coronary syndrome (ACS) patients, collected in two periods of time at 7 days and at 30 days post-ACS. MI patients were compared with those with UA (not healthy controls), thereby concentrating on variations in mRNA expression due to acute clinical conditions rather than underlying atherosclerosis and its treatment. Datasets were processed on the Affymetrix Human Genome U133 plus 2.0 platform. The robust multi-array averaging (RMA) method of the “affy” package in R was used to normalize the dataset.
Experimental: we used the open-source Anaconda Distribution to perform Python to apply feature selection and classification methods. Online converter was used to convert dataset format from text original format file into four excel files due to its large size, which was read as a single file in Python. The subjects of the Rothman dataset divided into testing and training sets to define the final classifier among all optimized lists. It proposed to classify MI patients from patients with unstable angina.
SVM classification methods was first applied to the data set without features selection method, we got only 6 genes and the classification accuracy was 91%. Revealed the effects of SVM classifier by applied 10-fold cross validation which result the overall accuracy of the model with append each score to a list and get the mean value -0.108. Then applied LOOCV to get more validation.
After that we compiled the information gain feature selection (IG) with decision tree method (DT). The IG method with decision tree algorithms were applied to the dataset using scikit learn package. Decision tree algorithm go throw selection best attribute using attribute selection measure (ASM) which (IG) measure. Then, spilt the dataset into smaller dataset with 7 genes and recursively for each child until one condition match the lowest accuracy. The classification. accuracy of information gain (IG) with decision tree (DT) algorithm was 100%. The result of this classification is overcoming the previous study as shown in the Table 3.
Table 3. Comparison between our proposed method and the most related work in literature base on classification accuracy and number genes
| Methods | Gene Selection Method | Classification Method | Dataset | Number of Genes | Classification Accuracy |
| Kazmi, et.al (2016) [8] | – P-Value
– t-tests |
– K-nearest neighbor (KNN) (k = 3).
Using (LOOCV) |
-Rothman
|
7
9 |
100%
88%. |
| Our Proposed Methods | Information gain (IG) | – Decision Tree (DT)
– SVM Using (LOOCV) |
-Rothman
|
6
7 |
91%
100% |
CONCLUSION
To conclude, bioinformatics analyzes were carried out on GSE29111’s microarray data set to explore CAD’s genetic mechanisms. This data set was previously used with different classification and selection methods than our research study, which improved the accuracy of classification by applying the decision tree, including the IG selection method, in addition to the SVM classification method. However, in future, further classification methods need to be applied in order to improve these findings.
REFERENCES
Cheng, M., An, S., & Li, J. (2017). CDKN2B-AS may indirectly regulate coronary artery disease-associated genes via targeting miR-92a. Gene, 629, 101-107.
Čulić, V., Eterović, D., Mirić, D., & Silić, N. (2002). Symptom presentation of acute myocardial infarction: influence of sex, age, and risk factors. American heart journal, 144(6), 1012-1017.
Gangwar, R. S., Rajagopalan, S., Natarajan, R., & Deiuliis, J. A. (2017). Noncoding RNAs in cardiovascular disease: pathological relevance and emerging role as biomarkers and therapeutics. American journal of hypertension, 31(2), 150-165.
Goldstein, J. A., Chinnaiyan, K. M., Abidov, A., Achenbach, S., Berman, D. S., Hayes, S. W., … & Shaw, L. J. (2011). The CT-STAT (coronary computed tomographic angiography for systematic triage of acute chest pain patients to treatment) trial. Journal of the American College of Cardiology, 58(14), 1414-1422.
Guo, S. Z., & Liu, W. J. (2019). Constructing differential co‑expression network to predict key pathways for myocardial infarction. Experimental and therapeutic medicine, 17(4), 3029-3034.
Jing Xu and Yuejin Yang (2021).Frontiers in Cardiovascular Medicine. Volume 8.https://doi.org/10.3389/fcvm.2021.736497
Kazmi, N., & Gaunt, T. R. (2016). Diagnosis of coronary heart diseases using gene expression profiling; stable coronary artery disease, cardiac ischemia with and without myocardial necrosis. PloS one, 11(3), e0149475.
Lu, Y., Meng, X., Wang, L., & Wang, X. (2018). Analysis of long non‑coding RNA expression profiles identifies functional lncRNAs associated with the progression of acute coronary syndromes. Experimental and therapeutic medicine, 15(2), 1376-1384.
Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., Cushman, M., … & Howard, V. J. (2016). Heart disease and stroke statistics-2016 update a report from the American Heart Association. Circulation, 133(4), e38-e48.
Slater, D. K., Hlatky, M. A., Mark, D. B., Harrell Jr, F. E., Pryor, D. B., & Califf, R. M. (1987). Outcome in suspected acute myocardial infarction with normal or minimally abnormal admission electrocardiographic findings. The American journal of cardiology, 60(10), 766-770.
Valensi, P., Lorgis, L., & Cottin, Y. (2011). Prevalence, incidence, predictive factors and prognosis of silent myocardial infarction: a review of the literature. Archives of cardiovascular diseases, 104(3),
Wu, K., Zhao, Q., Li, Z., Li, N., Xiao, Q., Li, X., & Zhao, Q. (2018). Bioinformatic screening for key mi RNA s and genes associated with myocardial infarction. FEBS open bio, 8(6), 897-913.
Zaiou, M., & Bakillah, A. (2018). Epigenetic regulation of ATP-Binding Cassette Protein A1 (ABCA1) gene expression: a new era to alleviate atherosclerotic cardiovascular disease. Diseases, 6(2), 34.

