1School of Biotechnology, Rajiv Gandhi Proudyogiki Vishwavidyalaya, Bhopal, Madhya Pradesh, India
2Department of Biological Sciences & Engineering, Maulana Azad National Institute of Technology, Bhopal, Madhya Pradesh, India
3Faculty of Computing and Information Technology, King Abdulaziz University, Rabigh, Kingdom of
Saudi Arabia
Article Publishing History
Received: 12/02/2016
Accepted After Revision: 22/03/2016
Coronaviruses such as SARS-CoV had caused high fatalness around the globe and have been a terror for public health, crafting a need for effective and active vaccines and drugs. Although this virus, coronavirus causing respiratory syndrome was controlled by non-vaccinated trials and measuresprimarily, still it persisted to be the leading threat for human health. Therefore thestrategy of optimal SAR-CoV drugs and vaccines is a priority, at present. Such drugs and vaccines represent key challenges: the immunity of coronavirus often diminishes quickly, entities craving to be sheltered include the elderly people, and vaccines may worsen rather than preventing coronavirus lung immunopathology. To address these issues, there is a need to promote the approach of prediction and then subjection. There is a necessity of that sort of drug or vaccine that could switch “ON” and “OFF” the metabolic pathways of an organism according to the requirement. Hence,the present study directs towards the prediction of novel drug targets that might aid in the treatment of Severe Acute Respiratory Syndrome and also in the treatment of Bovine Respiratory Disease. Complex.
Drugs; Immunity; Metabolic; Switch; Vaccines
Pradhan P, Shrivastava R, Tayubi I. A. The Unmasking of Un-Translated Regions of Human Coronavirus OC43 Against Severe Acute Respiratory Syndrome: An in Silico Study for Drug Targeting. Biosc.Biotech.Res.Comm. 2016;9(1).
Pradhan P, Shrivastava R, Tayubi I. A. The Unmasking of Un-Translated Regions of Human Coronavirus OC43 Against Severe Acute Respiratory Syndrome: An in Silico Study for Drug Targeting. Biosc.Biotech.Res.Comm. 2016;9(1). Available from: https://bit.ly/31bpG0m
Introduction
Severe acute respiratory syndrome-linked coronavirus (SARS-CoV) was acknowledged in the year 2003 when a sequence of deadly pneumonia cases emerged in Hong Kong (Drosten et al., 2003, Rota et al., 2003). SARS-CoV lung septicity has been characterized by a noticeable inflammatory and provocative cell infiltrate with severe alveolar damage (Nicholls et al., 2003, Nieto-Torres et al., 2014). Before the intensity of the disease was measured and controlled approximately eight thousand humans were clinically infected, with an inclusive case fatality rate of 10%, with mortality rate at 50%. This observation was emphasized on the population over the age of 65 years (Graham et al., 2013). It was also observed that after the salvage from coronavirus infections, previously infected entities may become susceptible to re-infection (Callow et al., 1990, Marra et al., 2003, Tang et al., 2011). In fact, those with declining immunity may be at high threat of even getting more severe effects upon the re-exposure of coronavirus (Nieto-Torres et al., 2014, Wang et al., 2014).
SARS-CoV is a positive-stranded RNA virus having length 29.7 kb with 14 open reading frames (ORF) approximately (Marra et al., 2003, Nieto-Torres et al., 2014). SARS-CoV grounds a respiratory ailment categorized by acute lung injury (ALI), and acute respiratory distress syndrome (ARDS). These extreme conditions can be described by diffuse alveolar damage, hyaline membrane formation, pulmonary cellular infiltration and edema accumulation, directing to hypoxemia and eventually to death (Ware and Matthay, 2000, Franks et al., 2003, Gralinski et al., 2013, Nieto-Torres et al., 2014, Honda-Okubo et al., 2015). SARS-CoV infection tempts migration of immune cells, such as neutrophils and macrophages to the lungs, initiating and amplifying the inflammation and chronic response (Lee et al., 2003, Yen et al., 2006, Darnell et al., 2007, Regla-Nava et al., 2015).
Another challenge for the inactivated SARS-CoV vaccine is the necessity forbiosafetylevel 3 (BSL3) amenities for the manufacture of vaccine. The very first SARS-CoV vaccine entrants were made from inactivated idled SARS-CoV. Inactivating the whole virus deprived of any adjuvant directed towards modest and unassertive protection, the only option to treat at the initial stage, was inducing low neutralizing-antibody titers and prior lung flushingthe perplexingferrets (Darnell et al., 2007, Honda-Okubo et al., 2015, Regla-Nava et al., 2015).
In mice, the inactive whole virus vaccines without or with combination (alum adjuvant) provided fractional protection, but this was accompanying the severe eosinophilic lung pathology (Bolles et al., 2011, Tseng et al., 2012, Honda-Okubo et al., 2015) similar to the pathology seen with SARS-CoV (Clay et al., 2012). To fight with these issues, a fruitful solution is the need of the hour so that a particular cure can be found out to fight the viral infections. These viral bodies contain un-translated regions that could aid in the development of inhibitors and drugs that could have a promising commercialization aspect.
As far molecular level is considered, it is a well-known fact thatwidespread study on RNA in contemporary times has stemmed into discovering a category of genetic regulatory element which is found in variety of organisms that too undergoing different folding and functional process leading to modification of gene and alteration in its expression, these regulatory elements are termed as riboswitch like elements (RLE) (Pradhan P et al., 2015). These elements are present have been acknowledged in the genomes of plants, archaea and fungi and are highly structured elements positioned in the 5’ un-translated region (UTR) of messenger RNA in the organism. The most vital characteristic of riboswitch is their folding into complex structures i.e. three dimensional structures (3D) to serve as a particular receptor for its target molecule that has its organization around the helical junctions along with the varying capacity and degrees of complexity. The aforementioned discussion has been used in bacterial metabolic pathways in the studies conducted (Mehta and Balaji, 2010).
This term “riboswitch” was coined by Dr. Ronald Breaker in the year 2002 when he reported that mRNA-encoding enzymes involved in vitamin B1 and B12 biosynthesis in E. coli could bind associated metabolites without the helper proteins being involved and they are RNA based components that can integrate ligand binding and gene regulation so as to dynamically respond to the molecular signals within cells (Winkler et al., 2002). These control several metabolic pathways counting the biosynthesis of vitamins and the breakdown of methionine, lysine and purines, comprising transcription cessation i.e. the termination process and initiation of translation (Eddy, 2001).A complex network of diverse inter metabolic pathways allowsthe organisms to consume nutrients from their surroundings to adapt to the environmental changes. Most of the well-characterized metabolic systems responding to external conditions are composed of proteins acting as sensors and/or regulators. However, variety of studies has shown that regulatory non-coding RNA structures in the untranslated region (UTR) play a crucial role in bacterial adaptive responses through sensing cellular metabolites (Bastet et al., 2011).
These riboswitches act similar to the “ON” and “OFF” mechanism of a switch, which could regulate the metabolism of the organism in a desired manner. Riboswitches are structurally divided in two parts:
- i) A sensor domain known as an aptamer which directly binds the small molecules and is evolutionary conserved domain in the UTR of an organism.
- ii) An expression platform thatgo throughits structural variations in response to the alterations sensed in, by the aptamer (Mandal and Breaker, 2004).
Riboswitches have a particulararray of ligand binding and its aptamer act in a way that the production of the metabolite generally proteins, gets terminated by self-destruction. Equating the diversity of riboswitches, taxonomically and regulatory molecular mechanisms it has triggered a focus that these switches represent the oldest regulatory systems (Blouin et al., 2009) among the organisms shown in figure 1.
 |
Figure 1: Riboswitches and their sensing capabilities (Barrick and Breaker, 2007, Regulski et al., 2008, Spinelli et al., 2008, Sudarsan et al., 2008, Henkin, 2009, Nechooshtan et al., 2009). |
Bacterial riboswitches devicevarious biological processes at numerousgoverning levels, such as transcription and translation (Waters and Storz, 2009). Collins et al., 2007 in their previous study had discussed that the glmS riboswitch perform its self-cleavage when bound to glucosamine 6-phosphate, commanding to rapid mRNA degradation (Collins et al., 2007). One of its own kind is a riboswitch that is a temperature-sensing riboswitch, known as thermo-sensor, which is subtle to the variation in temperature (Narberhaus et al., 2006). By and large, thermo-sensors block the translation at low temperature by confiscating the Shine–Dalgarno sequence and initiation codon sequence into a stem-loop structure. When there is a subsequent rise in temperature, the riboswitch assembly is denatured and translation signals are directed toribosomes to permitthe ribosome binding and translation initiation.
Thus, the folding of thermo-sensors is dependent on the temperature gradient experienced by an organism(Nocker et al., 2001, Johansson et al., 2002, Narberhaus et al., 2006, Rinnenthal et al., 2010). As far as genomic criteria are concerned, two remarkable properties of riboswitches set them apart from the majority of the other regulatory systems. Most importantfeature is the range of organisms, in which riboswitch like elements have been found. Most diverse is the distribution of THI-riboswitch like elements that has been witnessed in eubacteria, archaea and eukaryotes. Subsequent feature is that riboswitch like elements regulate a number of different processes; the most characteristic situation is the standard attenuation mechanism (Vitreschak et al., 2004).
Experimental Analysis
For the first time,riboswitch mechanism was decisivelyverified in a natural metabolic system in the year 2002 (Breaker, 2012). Ellington and Szostak stated an in vitro protocol that was dealing with the isolation of RNA aptamers which could bind to specific dyes, in 1990. The remarkable aspect about these studies is the creation of an RNA aptamer that did not necessitate a pre-existing RNA scaffold. Paralleled to most protein engineering energies, RNA aptamers can be madede novo by exposingnumerous randomized oligonucleotides to an in vitro selection process acknowledged as Systematic Evolution of Ligands by EXponential enrichment (SELEX). It requires two alternating steps of partitioning and amplifying the desired sequences of RNA aptamers, for its usage in the process.
Kinetic Control Of Riboswitches
Riboswitches can reversibly switch back and forth between an ‘ON’ and ‘OFF’ state, depending upon the concentration of the ligand that triggered the active site. When the aptitude to repress or activate the gene expression is dictated by its characteristic affinity for the ligand, a riboswitch like element is said to be thermodynamically regulated. In contrast, functional studies of a riboswitches have exposed that they entail a much higher ligand concentration to be activated, and in turn altering the level of gene expression of the organism. Evidently because riboswitches do not reach equilibrium with the ligand, the analogy is more like that of a fuse than a switch termed as “ribofuse” (Mehta and Balaji, 2010).In an in vitro study it has been discussed that riboswitch includes mRNA transcripts that can sense the concentration of metabolites over binding the target compound and then regulating the expression of the genes related with the metabolites responding to the concentration of metabolite [42]. Even though ample of riboswitch associated study has captivated on the significant capacity of various aptamer domains so as to adopt the complex structures of biological metabolites that are essential to bind with high affinity and specificity(Wickiser, 2009).
Material And Methods
The proposed framework for in silico prediction and identification of riboswitches, their ligands and its inhibitors are given below and also have been shown in figure 2.
 |
Figure 2: Flow chart of framework |
- Identification of riboswitch like gene sequence
- Riboswitch like gene sequence similarity searchbetween the desired organism’s nucleotide sequence and human nucleotide sequence (BLAST)
- Transcription (online) to corresponding Riboswitch like Element (RLE)
- 3D and 2D Prediction of RLE structure
- Molecular Docking- Blind Docking (BD)
- Molecular Docking- Focused Docking (FD) for the identification of ligand binding sites
- Enhancement of docking results
- Virtual screening using NCI diversity set by Raccoon-AutoDock
- Assessment of physico-chemical characters of lead compounds
- Prediction of ADME, toxicity and hydrophobicity of lead compounds
Methodology
Identification of riboswitch like gene sequence
The whole genome of Human coronavirus OC43 was acquired from the National Center for Biotechnology Information (NCBI) Gene Bank in FASTA format(Vijgen et al., 2005, Vijgen et al., 2015). The viral genome was searched for desired sequences present in its UTR by an online tool Riboswitch Explorer (RibEx) to identify the desired sequence termed as Riboswitch like sequences(Abreu-Goodger and Merino, 2005).
Riboswitch like gene sequence similarity search between the desired organism’s nucleotide sequence and human nucleotide sequence (BLAST)
BLAST utilized for the sequence similarity search. This tool let the user to know about the percentage of similarity between sequences. Sequences having more than 70-75% of the similarity are taken into account for further analysis and experimentation(Altschul et al., 1990).
Transcription (online) to corresponding Riboswitch Like Element (RLE)
Gene sequencesacknowledged by tool RibEx are transcripted to its parallel riboswitch by using the online “Transcription and Translation Tool” (Transcription and Translation Tool, 2015).
3D and 2D Prediction of RLE structure
Three dimensional(3D) structure of the RLE was anticipated by using an online program RNA fold (2D) (Vienna, 2015)and RNA Composer (2D to 3D)(Popenda et al., 2012). RNA Composer system offered a new user-friendly approach to fully automated prediction RNA 3D structures. This method was based on the machine translation principle and operated on the RNA FRABASE database acting as the dictionary, relating the RNA secondary structure and tertiary structure elements of riboswitch present in the complete genome of H. coronavirus OC43.
Molecular Docking- Blind Docking (BD)
In the present study, AutoDock based BDwasconducted to explorean apt binding site present in the predicted riboswitch like element of Human coronavirus OC43. In modus operandi of blind docking(VLifeMDS, 2011)whole structure of riboswitch is roofedunderneaththe virtual three dimensional grid box for the processing of docking. Coordinates of the grid box has been shown in Table 1. Altogetherthe computational studies were conceded out by means of AutoDock version 4.2 installed on a single machine whose configurationis 2.80 GHz Intel core2 duo processor using 3 GB Random Access Memory (RAM) and 320 GB hard disk alongwith Windows XP as an operating system.Docking software’s like “AutoDock”, “Autogrid” and “AutoDock version 4.2” were downloaded from the Scripps gateway. AutoDock software explored the entire surface of the 3D structure of riboswitch for binding of ligand in the cavities i.e. binding sites(Goodsell et al., 1996).
| Table 1: The coordinates of grid box for the Blind Docking (BD) | |||||||
| Target Protein | x-D | y-D | z-D | Spacing (Á) | x center | y center | z center |
| SEQ1 | 40 | 40 | 40 | 0.375 | 19.664 | -4.443 | -5.899 |
Alanine(ALA)was utilized as the ligand in blind docking technique so as to identify the binding sites present in the predicted riboswitch, as it is considered as the neutral and simplest ligand out of all the amino acids as a substrate for riboswitch like sequence.Particular binding sites existing in/on the surface of the predicted riboswitch like structurewereacknowledged on the foundation of lowest binding energy (-5 to -15 kcal/mol), so that the most stabilized structure can be retrieved. The particular binding site existing in/on the predicted riboswitch of H. coronavirus OC43 was used for the identification of binding residues involved in the present study. The identified binding residues present in the predicted riboswitchwere further utilized for the procedure of focused docking.
Focused Docking (FD) for the identification of ligand binding sites
Focused grid box casing ligand as well as binding residues involved in binding of ligand, wasarranged, precisely targeting specific ligand binding site for the recognized riboswitch of Humancoronavirus OC43 (Goodsell et al., 1996, Vijgen et al., 2015).
The X,Y and Z coordinates encompassing the data about the dimensionsof grid box in focused docking of anticipated Human coronavirus OC43 riboswitch has beentabularized in Table 2, these grid dimensions are further exploited for focused docking with different amino acids for the identification of specific receptor/substrate ligand interaction for the predicted riboswitch.
| Table 2: The coordinates of grid box for the Focused Docking (FD) | |||||||
| Target Protein | x-D | y-D | z-D | Spacing (Á) | x center | y center | z center |
| SEQ1 | 40 | 40 | 40 | 0.375 | 19.664 | -4.443 | -5.899 |
Enhancement of docking results
Subsequently, identifying a suitable ligand for the predicted riboswitch of Human coronavirus OC43 by the molecular modeling technique i.e. focused docking was accomplished. Distinct docking of the identified substrate ligand with its corresponding riboswitch,was done for the riboswitch identified in the genome of organism Humancoronavirus OC43 repetitively for a n-number of times for the improvement of the results(Goodsell et al., 1996, Vijgen et al., 2015).
Virtual screening using NCI diversity set by Raccoon-AutoDock
The identified binding sites present in predicted structure of novel Human coronavirus OC43 riboswitchis exploited for the virtual screening viaNCI Diversity Set containing diversity of drug molecules (Bikádi et al., 2006). Essential files for the process of virtual screening wereprimed by the online software Raccoon (Forli, 2010).
Raccoon has been used as a graphical user interface for AutoDock virtual screening. It can split multiple molecule ligand library files, convert them into the AutoDock format (i.e. *.pdbqt), and filter them by using common criteria (e.g., Lipinski’s rule of five, fragment-like “rule of three” and drug-likeness). A validation check of the input files wasaccomplished at every step, which comprises a check for the presence of non-standard atom types and ensuring that parameters, input file names, and grid maps have a coherent format.
Molecular docking simulation constructed virtual screening of Human coronavirus OC43 riboswitch was done using similar docking and grid parameters used in focused docking earlier. The coordinates of the grid box used in the virtual screening process of molecular libraries downloaded from ZINC database(Irwin and Shoichet, 2005) against the binding site of riboswitch (Human coronavirus OC43, SEQ1) has been tabulated in Table 3.
| Table 3: The coordinates of grid box for the virtual screening | |||||||
| Target Protein | x-D | y-D | z-D | Spacing (Á) | x center | y center | z center |
| SEQ1 | 40 | 40 | 40 | 0.375 | 19.664 | -4.443 | -5.899 |
Assessment of physico-chemical characters of lead compounds
Using zinc database, the best five marked off ligands for the riboswitch identified were evaluated for significant physicochemical properties such as xlog P, H-bond donors, H-bond acceptors, tPSA (Ų), molecular weight (g/mol), etc.
Prediction of ADME, toxicity and hydrophobicity of lead compounds
The top 5 lead molecules for Human coronavirus OC43 riboswitchweregauged using PreADMET online program(Kwang, 2005) for the check of toxicity and ADME properties (Hetényi and van der Spoel, 2002). This program device the presence of major toxicities such as mutagenicity, irritant effect, reproductive effects, etc. in the principle molecules on the basis of functional group present in their chemical conformation. PREADMET also calculates drug-likeness and drug score of principle molecules on the basis of their physico-chemical properties.Hydrophobicity of the lead molecules i.e. bio-molecular complexes were detected with the aid of PLATINUM (Protein-Ligand ATtractions Investigation NUMerically) web-service. It estimates the hydrogen bonding interactions among the bio-molecular complexes, stacking interactions and hydrophobic interactions as well. This feature of PLATINUM can be utilized to screen docking processes conducted by AutoDock, GOLD, etc. This program generates 2D hydrophobicity maps for lipid bilayers and helical peptides. This does not aid in the process of docking, it only retrieve 3 dimensional coordinates of the biological molecules (ligand and receptors) to compute and envisage the hydrophobic/hydrophilic properties between them (Pyrkov et al., 2009).
Results And Discussion
The stepwise results obtained are summarized and discussed below:
Exploration of the genome assembly for identification of riboswitch like sequence:
The following sequence was identified for transcription into riboswitch like elements present in the viral DNA of Human coronavirus OC43 causing Severe Acute Respiratory Syndrome.
Sequence similarity with human genome: BLAST Score
The identified riboswitch like sequence of H. coronavirus OC43 had shownfollowing results on NCBI database search for gene sequence similarity present in the genome of Homo sapiens.
SEQ1-tgctaGTCTTGTTCTGGCAAAACTTGGCAAGGATgccac
This gene sequence from Human coronavirus OC43 having the identified riboswitch had shown a BLAST score 77.8 with a maximum 100% of gene sequence similarity with Bovine coronavirus strain. The BLAST result of human gene sequence similarity to SEQ1 gene sequence is shown in figure 4, depicting no gene sequence similarity.
 |
Figure 3: Riboswitch like sequence |
 |
Figure 4: BLAST between Human coronavirus and Homo sapiens |
Thus, human genome does not have any similarity with the Human coronavirus OC43 directing towards non-presence of any riboswitch like gene sequence as predicted by RibEx.
Transcription of gene sequence to RLE
Sequence after transcription:
SEQ1-GUUCUGGCAAAACUUGGCAAGGAUGCCAC
The acknowledged gene sequencewas transcripted to its corresponding riboswitch like elements using “Transcription and Translation Tool”. This tool interchanges the thymine (T) with the nucleotide uracil (U) to form its equivalent riboswitch.
Predicted tertiary structures of riboswitch
Tertiary structure of the identified riboswitch in Human coronavirus OC43 were predicted by online program RNAfold(2D)(Vienna, 2015)and RNAcomposer (2Dto 3D)(Popenda et al., 2012), SEQ1 riboswitch consists of a single chain of 39 nucleotides. The predicted secondary and tertiary structure of SEQ1 riboswitch in H. coronavirus OC43 has been shown belowin Figure 5a (2D) and 5b (3D).
 |
Figure 5a |
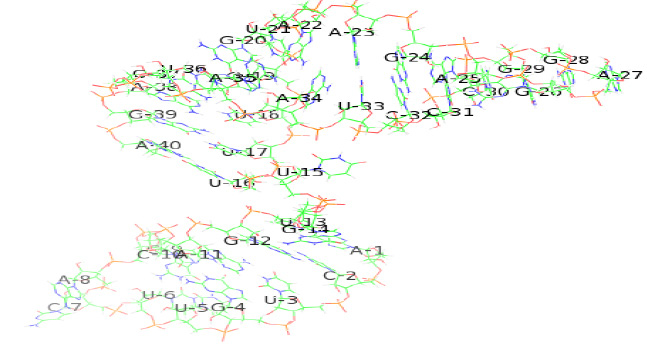 |
Figure 5b |
Blind and Focused Docking results
Binding site present in H. coronavirus riboswitch SEQ1 identified by FD with LYS suggested that there are some residues ingene sequence that are involved and are capable in ligand binding.Docking results obtained by BD of the riboswitch i.e. SEQ1 has been tabulated in Table 4.The results of FD of SEQ1 using the identified binding residues intricate in the binding of the ligand to the predicted riboswitch advocated that the amino acid lysine (LYS) as its substrate ligand had displayed the superlative binding amongstthe 20 amino acids with the Binding Energy (BE) value as -5.70 kcal/mol (Ki = 66.22 μM) that has beentabulated in Table 5.
| Table 4: Blind docking results | |||
| S. No. | Ligand | Binding Energy (kcal/mol) | Ki (μM) |
| SEQ1 | GLY | -3.79 | 1.66 |
| Table 5: Focused docking results for SEQ1 | ||
| Amino acid | BE (kcal/mol) | Ki (µM) |
| ALA | -4.30 | 701.14 |
| ARG | -4.34 | 22.68 |
| ASN | -4.66 | 380.87 |
| ASP | -2.18 | 25.1 |
| CYS | -5.50 | 92.58 |
| GLN | -4.52 | 489.94 |
| GLU | -.170 | 138.38 |
| GLY | -3.85 | 1.52 |
| HIS | -.350 | 648.24 |
| ILE | -4.04 | 1.08 |
| LEU | -4.54 | 473.4 |
| LYS | -5.70 | 66.22 |
| MET | -4.66 | 382.83 |
| PHE | -5.48 | 99.77 |
| PRO | -3.89 | 1.42 |
| SER | -.430 | 3.07 |
| THR | -3.91 | 1.36 |
| TRP | -5.02 | 209.41 |
| TYR | -4.88 | 266 |
| VAL | -4.41 | 583.85 |
Further, to enhance the rate of affinity, the results of individual docking of riboswitch SEQ1 with LYS for a repeated number of timeswas conducted to enhance the affinity of receptor and ligand for which the result has been shown in Table 6.
| Table 6: Enhanced docking results | |||
| Riboswitch | Ligand | BE (kcal/mol) | Ki (μM) |
| SEQ1 | LYS | -7.01 | 7.26 |
Virtual Screening results
The 5 lead/principle molecules were selected after virtual screening against predicted Human coronavirus OC43 riboswitch of SEQ1. The BE and Ki value of 5 principle molecules for the H. coronavirus OC43 riboswitch has beentabulated in Table 7.
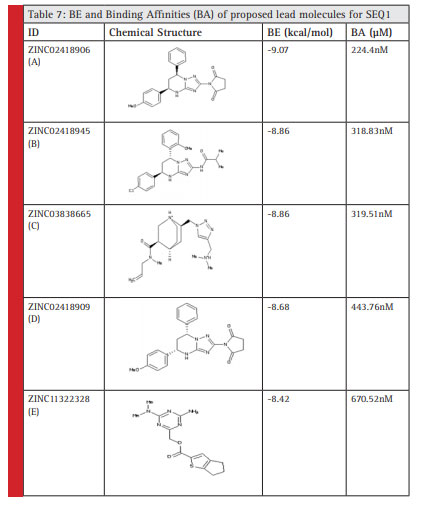 |
Table 7 |
The following five lead molecules were obtained for SEQ1 riboswitch after the virtual screening; these were obtained from ZINC database: ZINC02418906 (A), ZINC02418945 (B), ZINC03838665 (C), ZINC02418909 (D)and ZINC11322328 (E).
Physico-chemical properties of screened lead compounds
The physico-chemical properties of top 5 lead molecules screened for SEQ1 Human coronavirus OC43 ribos witch has been tabulated in Table 8.
| Table 8: Physio-chemical parameters of proposed lead molecules for SEQ1 | ||||||
| Parameter | Optimum range | A | B | C | D | E |
| xlogP | -5 to +5 | 1.53 | 4.24 | 0.91 | 1.53 | 2.15 |
| H-bond donors | 0 to 5 | 1 | 2 | 2 | 1 | 2 |
| H-bond acceptors | 0 to 10 | 1 | 7 | 7 | 8 | 7 |
| tPSA(Ų) | 60 to 140 | 89 | 81 | 59 | 89 | 94 |
| Molecular weight (g/mol) | 150 to 500 | 403.442 | 425.92 | 348.495 | 403.442 | 319.39 |
ADME &Drug-likeness profiling
The results of drug-likeness and ADME prediction of selected top 5 lead compounds for the predicted riboswitch has been worked out by PreADMET online program. The ADME and drug likeness score for top 5 virtually screened lead molecule for SEQ1 riboswitch has been shown in Table 9-10 respectively. The two lead compounds out of top 5 screened leads ZINC02418945 (B), ZINC11322328 (E)passed the ADMET with a good drug likeness score.
| Table 9: ADME results | |||||
| Parameter | A | B | C | D | E |
| BBB (blood brain barrier) | 0.208578 | 0.886191 | 0.089131 | 0.208578 | 0.112125 |
| CaCO2 | 22.277 | 27.3611 | 27.0288 | 22.277 | 11.6979 |
| HIA (Human Intestinal Absorption) | 97.202625 | 95.22717 | 86.59872 | 97.20262 | 89.00412 |
| Plasma_Protein_Binding | Inhibitor | 89.69697 | 6.966986 | Inhibitor | 86.29595 |
| Skin_Permeability | -3.74703 | -3.19318 | -4.54874 | -3.74703 | -4.22118 |
| Table 10: Drug-likeness results | |||||
| Parameter | A | B | C | D | E |
| CMC_like_Rule | Qualified | Qualified | Failed | Qualified | Qualified |
| CMC_like_Rule_Violations | 0 | 0 | 1 | 0 | 0 |
| Lead_like_Rule | Violated | Violated | Violated | Violated | Suitable |
| MDDR_like_Rule | Mid-structure | Mid-structure | Drug-like | Mid-structure | Mid-structure |
| Rule_of_Five | Suitable | Suitable | Suitable | Suitable | Suitable |
| Rule_of_Five_Violations | 0 | 0 | 0 | 0 | 0 |
| WDI_like_Rule | In 90% cutoff | In 90% cutoff | Failed | In 90% cutoff | In 90% cutoff |
| Table 11: Hydrophobicity results | |||||
| Bonding | Ligand | ||||
| A | B | C | D | E | |
| H-bonds | 0.01 | 0.80 | 0.01 | 0.01 | 0.04 |
| S(L/L) | 0.06 | 0.00 | 0.00 | 0.19 | 0.00 |
| S(H/H) | 102.15 | 55.38 | 66.39 | 38.10 | 96.96 |
| Sburied | 204.93 | 192.21 | 113.67 | 250.25 | 169.43 |
| Stotal | 348.59 | 366.58 | 333.29 | 347.34 | 299.68 |
| Match1 | 0.2932 | 0.1511 | 0.1992 | 0.1102 | 0.3225 |
| Match2 | 0.0005 | 0 | 0 | 0.0017 | 0 |
| Stack | 1.97 | 0.24 | 0 | 2.79 | 1.62 |
 |
Figure6: Biinding sites (A29, A30) predicted after focused docking by PyMol |
 |
Figure 7: 2D-Hydrophobicity Map: Brown-Cyan color–brown (oil-colored) hydrophobic and cyan (water-colored) hydrophilic properties(Pyrkov et al., 2009) |
Hydrophobicity/Hydrophilicity
PLATINUM program was used for the detection of hydrophobicity/hydrophilicity of the ligands. Most of the ligands were found to be hydrophilic in nature. The interactions between the ligands and other molecules are principally electrostatic in nature as these objects shows polarity. Hydrophilic interaction is in force not only in solvating ions, but also in stabilizing DNA, proteins, etc. As per the tutorial of PLATINUM program, an hydrogen bond has been assigned a value ranging from 0.0-1.0, 1.0 being the perfect geometry, so ligand B (ZINC02418945) is near the range of perfect geometry, aiding in the stability of the ligand-receptor complex.
Conclusion
We have been able to accomplish in silico prediction and identification of the riboswitch for Human coronavirus OC43 straincausing Severe Acute Respiratory Syndrome (SARS). This approach has given supplementary information about the peculiar binding sites and ligands for the strain of H. coronavirus OC43. After the comprehensive analysis of molecular docking study this prediction approach has drawn our focus that the complete genome of Human coronavirus OC43 is having elements that might act as switches for the “ON” and “OFF” mechanisms of metabolic pathway involved in the detrimental viral activities of an organism. The stretch of sequence (RLE) obtained after the analysis has paved the way for the receptor-ligand interaction, in which the potent ligand found was LYS amino acid that has the leading affinity for the binding site present on the surface of the receptor (RLE). This ligand has shown its competency amongst 20 different amino acids. Further, the binding sites: A29 and A30 can accommodate the structure of the ligand in a definite manner, as per the conclusions drawn after focused docking. The predicted RLEis having its vital role in the functioning of gene encoding nucleocapsid protein that directs towards targeting the encoded protein so as to regulate the activities of the organism inside humans. Also, the sequence obtained does not have any similarity with the genome of humans, which shows that the inhibitors/lead compounds retrieved from the ZINC database will not hinder the metabolic pathway of humans and their metabolic activities, if the organism is targeted inside their body. These inhibitors anticipated are free from the side effects of the antiviral drugs and have distant chances of emerging resistance in humans. Furthermore,targeting nucleocapsid protein has also proven its authenticity after the similarity search via BLAST since the predicted sequence has its root in Bovine coronavirus also, that has been acting as the major causing agent to the members of subfamily Bovinae. This subfamily is having a diversified group of 10 genera that are the victims of this organism and are prone to respiratory and enteric infections.Hence, the study directs towards the prediction of novel drug targets that might aid in the treatment of Severe Acute Respiratory Syndrome and also Bovine Respiratory Disease Complex.
Future Prospects
Further, aptamers and the RNA structures can be generated by in vitro selection to interact with small molecules for the expression and binding of a wide range of ligands (Famulok, 1999, Hermann and Patel, 2000).
Moreover, riboswitches can also be worked on in vivo experiments as an artificial regulatory construct that involves an aptamer structure leading to the addition of the aptamer-binding site for a compound stabilizing a hairpin structure that interfered with initiation of translation of a reporter gene (Werstuck and Green, 1998). It is also possible to use aptamers for the regulation of cellular processes, such as the cell cycle (Grate and Wilson, 2001).
References
Abreu-Goodger, C. &Merino, E. (2005). RibEx: a web server for locating riboswitches and other conserved bacterial regulatory elements. Nucleic acids research, 33, W690-W692.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J.(1990.) Basic local alignment search tool. Journal of molecular biology, 215, 403-410.
Barrick, J. E. &Breaker, R. R. (2007). The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome biology, 8, R239.
Bastet, L., Dubé, A., Massé, E. &Lafontaine, D. A. (2011). New insights into riboswitch regulation mechanisms. Molecular microbiology, 80, 1148-1154.
Bikádi, Z., Hazai, E., Zsila, F. &Lockwood, S. F. (2006). Molecular modeling of non-covalent binding of homochiral (3S, 3´ S)-astaxanthin to matrix metalloproteinase-13 (MMP-13). Bioorganic & medicinal chemistry, 14, 5451-5458.
Blouin, S., Mulhbacher, J., Penedo, J. C. &Lafontaine, D. A. (2009.) Riboswitches: ancient and promising genetic regulators. Chembiochem, 10, 400-416.
Bolles, M., Deming, D., Long, K., Agnihothram, S., Whitmore, A., Ferris, M., Funkhouser, W., Gralinski, L., Totura, A. &Heise, M. (2011). A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. Journal of virology, 85, 12201-12215.
Breaker, R. R.(2012). Riboswitches and the RNA world. Cold Spring Harbor perspectives in biology, 4, a003566.
Callow, K., Parry, H., Sergeant, M. &Tyrrell, D. (1990). The time course of the immune response to experimental coronavirus infection of man. Epidemiology and infection, 105, 435-446.
Clay, C., Donart, N., Fomukong, N., Knight, J. B., Lei, W., Price, L., Hahn, F., Van Westrienen, J. &Harrod, K. S. (2012). Primary severe acute respiratory syndrome coronavirus infection limits replication but not lung inflammation upon homologous rechallenge. Journal of virology, 86, 4234-4244.
Collins, J. A., Irnov, I., Baker, S. &Winkler, W. C. (2007.) Mechanism of mRNA destabilization by the glmS ribozyme. Genes & development, 21, 3356-3368.
Darnell, M. E., Plant, E. P., Watanabe, H., Byrum, R., Claire, M. S., Ward, J. M. & Taylor, D. R. (2007). Severe acute respiratory syndrome coronavirus infection in vaccinated ferrets. Journal of Infectious Diseases, 196, 1329-1338.
Drosten, C., Günther, S., Preiser, W., Van Der Werf, S., Brodt, H.-R., Becker, S., Rabenau, H., Panning, M., Kolesnikova, L. & Fouchier, R. A. (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New England Journal of Medicine, 348, 1967-1976.
Eddy, S. R. (2001). Non–coding RNA genes and the modern RNA world. Nature Reviews Genetics, 2, 919-929.
Famulok, M. (1999.) Oligonucleotide aptamers that recognize small molecules. Current opinion in structural biology, 9, 324-329.
Forli, S. (2010.) Raccoon| AutoDock VS: an automated tool for preparing AutoDock virtual screenings.
Franks, T. J., Chong, P. Y., Chui, P., Galvin, J. R., Lourens, R. M., Reid, A. H., Selbs, E., Mcevoy, C. P. L., Hayden, C. D. L. & Fukuoka, J. (2003). Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Human pathology, 34, 743-748.
Goodsell, D. S., Morris, G. M. & Olson, A. J. (1996) Automated docking of flexible ligands: applications of AutoDock. Journal of Molecular Recognition, 9, 1-5.
Graham, R. L., Donaldson, E. F. & Baric, R. S. (2013). A decade after SARS: strategies for controlling emerging coronaviruses. Nature Reviews Microbiology, 11, 836-848.
Gralinski, L. E., Bankhead, A., Jeng, S., Menachery, V. D., Proll, S., Belisle, S. E., Matzke, M., Webb-Robertson, B.-J. M., Luna, M. L. & Shukla, A. K. (2013). Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. MBio, 4, e00271-13.
Grate, D. & Wilson, C. (2001). Inducible regulation of the S. cerevisiae cell cycle mediated by an RNA aptamer–ligand complex. Bioorganic & medicinal chemistry, 9, 2565-2570.
Henkin, T. M. (2009). RNA-dependent RNA switches in bacteria. Riboswitches. Springer.
Hermann, T. & Patel, D. J. (2000). Adaptive recognition by nucleic acid aptamers. Science, 287, 820-825.
Hetényi, C. & Van Der Spoel, D. (2002). Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein science, 11, 1729-1737.
Honda-Okubo, Y., Barnard, D., Ong, C. H., Peng, B.-H., Tseng, C.-T. K. & Petrovsky, N. (2015). Severe Acute Respiratory Syndrome-Associated Coronavirus Vaccines Formulated with Delta Inulin Adjuvants Provide Enhanced Protection while Ameliorating Lung Eosinophilic Immunopathology. Journal of virology, 89, 2995-3007.
Irwin, J. J. & Shoichet, B. K. (2005). ZINC-a free database of commercially available compounds for virtual screening. Journal of chemical information and modeling, 45, 177-182.
Johansson, J., Mandin, P., Renzoni, A., Chiaruttini, C., Springer, M. & Cossart, P. (2002). An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell, 110, 551-561.
Kwang, L. S.(2005) In silico high-throughput screening for ADME/Tox properties: PreADMET program. Abstr Conf Comb Chem Jpn, 2005. 22-28.
Lee, N., Hui, D., Wu, A., Chan, P., Cameron, P., Joynt, G. M., Ahuja, A., Yung, M. Y., Leung, C. & To, K. (2003). A major outbreak of severe acute respiratory syndrome in Hong Kong. New England Journal of Medicine, 348, 1986-1994.
Mandal, M. & Breaker, R. R. (2004). Gene regulation by riboswitches. Nature Reviews Molecular Cell Biology, 5, 451-463.
Marra, M. A., Jones, S. J., Astell, C. R., Holt, R. A., Brooks-Wilson, A., Butterfield, Y. S., Khattra, J., Asano, J. K., Barber, S. A. & Chan, S. Y. (2003). The genome sequence of the SARS-associated coronavirus. Science, 300, 1399-1404.
Mehta, N. B. & Balaji, P. (2010). Riboswitches: classification, function and insilico approach. Int J, 1, 409-420.
Narberhaus, F., Waldminghaus, T. & Chowdhury, S. (2006). RNA thermometers. FEMS microbiology reviews, 30, 3-16.
Nechooshtan, G., Elgrably-Weiss, M., Sheaffer, A., Westhof, E. & Altuvia, S. 2009. A pH-responsive riboregulator. Genes & development, 23, 2650-2662.
Nicholls, J., Dong, X. P., Jiang, G. & Peiris, M. (2003) SARS: clinical virology and pathogenesis. Respirology, 8, S6-S8.
Nieto-Torres, J. L., Dediego, M. L., Verdiá-Báguena, C., Jimenez-Guardeño, J. M., Regla-Nava, J. A., Fernandez-Delgado, R., Castaño-Rodriguez, C., Alcaraz, A., Torres, J. & Aguilella, V. M. (2014.) Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog, 10, e1004077.
Nocker, A., Hausherr, T., Balsiger, S., Krstulovic, N.-P., Hennecke, H. & Narberhaus, F. (2001). A mRNA-based thermosensor controls expression of rhizobial heat shock genes. Nucleic acids research, 29, 4800-4807.
Popenda, M., Szachniuk, M., Antczak, M., Purzycka, K. J., Lukasiak, P., Bartol, N., Blazewicz, J. & Adamiak, R. W. (2012). Automated 3D structure composition for large RNAs. Nucleic acids research, gks339.
Pradhan P, Soni N. K, Chaudhary L, Mujwar S & R., P. K. (2015). In-Silico Prediction of Riboswitches and Design of their Potent Inhibitors for H1N1, H2N2 and H3N2 Strains of Influenza Virus. Biosci Biotechnol Res Asia, 12, 3.
Pyrkov, T. V., Chugunov, A. O., Krylov, N. A., Nolde, D. E. & Efremov, R. G. (2009). PLATINUM: a web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes. Bioinformatics, 25, 1201-1202.
Regla-Nava, J. A., Nieto-Torres, J. L., Jimenez-Guardeño, J. M., Fernandez-Delgado, R., Fett, C., Castaño-Rodríguez, C., Perlman, S., Enjuanes, L. & Dediego, M. L. (2015). Severe Acute Respiratory Syndrome Coronaviruses with Mutations in the E Protein Are Attenuated and Promising Vaccine Candidates. Journal of virology, 89, 3870-3887.
Regulski, E. E., Moy, R. H., Weinberg, Z., Barrick, J. E., Yao, Z., Ruzzo, W. L. & Breaker, R. R. (2008). A widespread riboswitch candidate that controls bacterial genes involved in molybdenum cofactor and tungsten cofactor metabolism. Molecular microbiology, 68, 918-932.
Rinnenthal, J., Klinkert, B., Narberhaus, F. & Schwalbe, H. (2010). Direct observation of the temperature-induced melting process of the Salmonella fourU RNA thermometer at base-pair resolution. Nucleic acids research, 38, 3834-3847.
Rota, P. A., Oberste, M. S., Monroe, S. S., Nix, W. A., Campagnoli, R., Icenogle, J. P., Penaranda, S., Bankamp, B., Maher, K. & Chen, M.-H. (2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science, 300, 1394-1399.
Spinelli, S. V., Pontel, L. B., Véscovi, E. G. & Soncini, F. C. (2008). Regulation of magnesium homeostasis in Salmonella: Mg2+ targets the mgtA transcript for degradation by RNase E. FEMS microbiology letters, 280, 226-234.
Sudarsan, N., Lee, E., Weinberg, Z., Moy, R., Kim, J., Link, K. & Breaker, R. (2008). Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science, 321, 411-413.
Tang, F., Quan, Y., Xin, Z.-T., Wrammert, J., Ma, M.-J., Lv, H., Wang, T.-B., Yang, H., Richardus, J. H. & Liu, W. (2011). Lack of peripheral memory B cell responses in recovered patients with severe acute respiratory syndrome: a six-year follow-up study. The Journal of Immunology, 186, 7264-7268.
Transcription and Translation Tool [Online]. Available: 2015 http://www.attotron.com/cybertory/analysis/trans.htm./
Tseng, C.-T., Sbrana, E., Iwata-Yoshikawa, N., Newman, P. C., Garron, T., Atmar, R. L., Peters, C. J. & Couch, R. B. (2012). Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS One, 7, e35421.
Using Vlifemds, M. D. (2011). VLife Product Documentation, Tutorial: BioPredicta, VLife Sciences Technologies Pvt. Ltd.
Vienna, R. (2015). Secondary Structure Prediction http://rna. tbi. univie. ac. at/cgi-bin. RNAfold. cgi.
Vijgen, L., Keyaerts, E., Moës, E., Thoelen, I., Wollants, E., Lemey, P., Vandamme, A.-M. & Van Ranst, M. (2005) Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. Journal of virology, 79, 1595-1604.
Vijgen, L., Keyaerts, E., Moes, E., Thoelen, I., Wollants, E., Lemey, P., Vandamme, A. M. & Van Ranst, M. 2015. Human coronavirus OC43, complete genome.
Vitreschak, A. G., Rodionov, D. A., Mironov, A. A. & Gelfand, M. S. (2004). Riboswitches: the oldest mechanism for the regulation of gene expression? TRENDS in Genetics, 20,44-50.
Wang, S.-F., Tseng, S.-P., Yen, C.-H., Yang, J.-Y., Tsao, C.-H., Shen, C.-W., Chen, K.-H., Liu, F.-T., Liu, W.-T. & Chen, Y.-M. A. (2014). Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochemical and biophysical research communications, 451, 208-214.
Ware, L. B. & Matthay, M. A. (2000). The acute respiratory distress syndrome. New England Journal of Medicine, 342, 1334-1349.
Waters, L. S. & Storz, G. (2009). Regulatory RNAs in bacteria. Cell, 136, 615-628.
Werstuck, G. & Green, M. R. (1998) Controlling gene expression in living cells through small molecule-RNA interactions. Science, 282, 296-298.
Wickiser, J. K. (2009) Kinetics of riboswitch regulation studied by in vitro transcription. Riboswitches: Methods and Protocols, 53-63.
Winkler, W. C., Cohen-Chalamish, S. & Breaker, R. R. (2002). An mRNA structure that controls gene expression by binding FMN. Proceedings of the National Academy of Sciences, 99, 15908-15913.
Yen, Y.-T., Liao, F., Hsiao, C.-H., Kao, C.-L., Chen, Y.-C. & Wu-Hsieh, B. A. (2006). Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. Journal of virology, 80, 2684-2693.

