1Centre for Drug Discovery and Development, Sathyabama Institute of Science & -Technology, Chennai – 600119, India
2 Department of Bioinformatics, Sathyabama Institute of Science & -Technology, Chennai – 600119,India
3 Sathyabama Institute of Science & -Technology, Chennai – 600119
4 California University of Science & Medicine, School of Medicine, CA, USA
5 Musculoskeletal Disease Research Laboratory US Department of Veteran Affairs, Loma Linda, CA, USA
Corresponding author email: jerrine.jj@gmail.com
Article Publishing History
Received: 17/04/2020
Accepted After Revision: 23/06/2020
Breast cancer is the most frequent cancer among women. The present study focuses on the comparative in-silico based investigation of the plant natural compounds namely Cubenin, Curcumin, Delta Cadiene, Eugenol, Terpene and Quercetin binding efficacy towards HER2 and their intermolecular interactions were compared with the commercial drugs:Cyclophosphamide, Doxorubin, Letrozole, Methotrexate and Tamoxifen.The comparative molecular docking was performed with the natural compounds and the synthetic drugs used in breast cancer treatment against the target HER2. The molecular docking analysis was done using Discovery Studio. The ADME properties were also studied. The observation of the common binding site for all the ligands confirms the binding pocket; where the isolated compound agrees well with the binding residues and thus can be optimized further to arrive at a molecule that has a high binding affinity and low binding constant. The results of the docking studies carried out on HER2 provide an insight for the natural compounds having druggable properties. These results are supportive to confirm the natural compounds from plants as a better lead for cancer therapeutics. Thus, the results of the docking studies carried out on HER2 corroborate to the findings that the most suitable drug like properties are possessed by the compound. In comparison with other compounds natural compounds are better and it is druggable. This provides evidence of how a natural compounds from plants can be a source of potential anti- cancer agent. The preclinical studies will pave way for a potential anti-cancer compound.
Docking; ADME; HER2; Discovery Studio
Deborah S. K, Raghavi R, Joseph J, Christy J, Aruni W. Insilico Docking Studies of Phytomolecules As Anti-Breast Cancer Agents. Biosc.Biotech.Res.Comm. 2020;13(2).
Deborah S. K, Raghavi R, Joseph J, Christy J, Aruni W. Insilico Docking Studies of Phytomolecules As Anti-Breast Cancer Agents. Biosc.Biotech.Res.Comm. 2020;13(2). Available from: https://bit.ly/2NawoPp
Copyright © Deborah et al., This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) https://creativecommns.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
Cancer is the leading causes of death globally where 1 in 6 die. Low economic prosperity and lack of awareness of prevention strategies are noted to be vital factors contributing to the burden and incidence for cancer ERBB is abbreviated from erythroblastic oncogene B, a gene isolated from avian genome. Heterodimerization of this receptor with other members of the EGFR family, typically owing to HER2 overexpression, results in the autophosphorylation of tyrosine residues within the cytoplasmic domain of the heterodimer and initiates a variety of signalling pathways leading to cellular proliferation and tumorigenesis (Yarden, et al.,2001) ).
Amplification or over-expression of this oncogene has been shown to play an important role in the development and progression of certain aggressive types of breast cancer. According to TCGA (The cancer Genome Atlas) data portal HER2 aberration studied in various solid tumours associated with cervical, bladder and were have limited therapeutic options (Cancer Genome Atlas,2013). Approximately, 70% of death due to cancer occurs in low- and middle-income countries. Receptor tyrosine-protein kinase erbB-2, otherwise known as HER2, a significant member in the EGFR family of receptor tyrosine kinases family, (Pegram et al.,2020 Siegel et al.,2020).
So, outspreading the HER2-based therapeutic option beyond breast cancer to other solid tumours with HER2 overexpression will be beneficial to the affected individuals. In recent years, the protein has become an important biomarker and target of therapy for approximately 30% of breast cancer patients. In this study, the natural compounds from plant source is considered based on their pharmacological properties. The compounds have been characterized in detail for breast cancer. The natural source would overcome the existing synthetic drugs in mode of action and also reduce the side effects caused by the commercial compounds (Christy and Swetha 2019).
The present study focuses on the comparative in-silico based investigation of the plant natural compounds namely Cubenin, Curcumin, Delta Cadiene, Eugenol, Terpene and Quercetin binding efficacy towards HER2 and their intermolecular interactions were compared with the commercial drugs:Cyclophosphamide, Doxorubin, Letrozole, Methotrexate and Tamoxifen. The in-silico approach enables one to screen for ADMET properties of vast number of molecules within short span thus reducing the time and is a non- expensive and non-tedious process with great accuracy, which is not possible in standard experimental methods.
MATERIAL AND METHODS
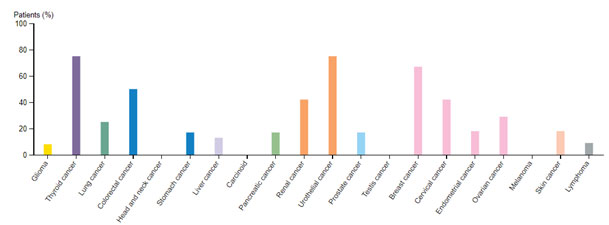
HER2/ERBB2 expression datamining: The cancer genome data repository was used to assess the HER2 positive cancer types and the Figure 1 revealed that HER2 expression was seen in other solid tumors associated with bladder, uterine, cervical regions also (TCGA GDC portal).Human pathology atlas module of human protein atlas also revealed the HER2 varied expression with breast, colorectal, cervical, renal and urothelial cancers and dataset related information’s listed in Figure 2. (Uhlen et al.,2017)
Figure 1: HER2 aberrations in various cancer types
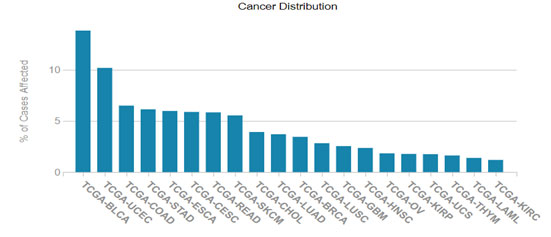
Figure 2: HER2 profile based on human pathology atlas
HER2 protein structure quality assessment:
The target is retrieved from the Protein Data Bank, RCSB-PDB (H.M Berman et al.,2000). Upon searching the protein structure for the HER2 protein, the structure 3RCD has been chosen the fitting protein model (Ishikawa et al.,2011). Stereochemical properties based assessment revealed that nearly 90% of residues are residing in the allowed region( ).Before proceeding with the docking the protein structure has been energy minimized to find an arrangement in space of a collection of atoms where, the net inter-atomic force on each atom is acceptably close to zero and the position on the potential energy surface (PES) is a stationary point . the energy value of the protein molecule after Energy minimization is-44093.590. The energy minimisation was done in Swiss PDB viewer, it is a downloadable software (Swiss PDB viewer). Then after this the protein structure was uploaded in the Discovery Studio, where the force field CHARMM was applied and then receptor binding pockets where determined so as the ligand fits in.
Preparation of Ligand: The commercial compounds Cyclophosphamide, Doxorubin, Letrozole, Methotrexate and Tamoxifen and the natural compounds Cubenin, Curcumin, Delta Cadiene, Eugenol, Terpene and Quercetin three dimensional structures were retrieved from Pubchem database (Kim et al.,2016). Then the ligands were analysed if they are druggable or not using the online tool, which is based on the Lipinski’s rule of 5 ( Jayaraman et al.,2012).
ADMET Profiling:The ADME profiling was done using the Accelrys Discovery Studio 2.5 software. The lead compounds from natural resources fail to enter the market due to the poor pharmacokinetic properties. So, designing ligands satisfying the Adsorption, Distribution, Metabolism Elimination and Toxicity (ADMET) properties will go through the market as a good drug. The drugs should be orally absorbed and distributed to the site of action and eliminated from the body without leaving any traces, which produces adverse effects. Hence, the tools and computer-aided methods, nowadays, have become popular in identifying good drug candidate molecule.
Molecular Docking: Molecular docking is a key tool in structural molecular biology and computer-assisted drug design (Venkatachalam et al.,2003). The goal of ligand-protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known three-dimensional structure. The receptor-ligand interaction uses Ligand Fit protocol, which docks the ligands into the binding site of receptors using shape-based searching and Monte Carlo sampling of ligands (Gangopadhyay et al,2017).
The parameters used are PLP1 algorithm for energy grid and conjugate gradient for energy minimization. The scores for docked poses are obtained by LigScore1, LigScore which predicts the binding affinities, -PLP1, -PLP2 known as Piecewise Linear Potential scoring function calculates both the shape and hydrogen bond complementarity of poses to the active site and Jain scoring function which scores the non-covalent protein-ligand interactions( Krammer et al.,2005;Jain 1996).
RESULTS AND DISCUSSION
The protein structure retrieved from RCSB-PDB: There were eight druggable cavities populated in the chains of HER2 three-dimensional structure and were ranked based on the cavity score and drug score. In general The ligandability score used to assess the possible small ligands affinity to the specified cavity, whereas druggability scores reveal and rank the good target for HER2 binding .Specified caities of HER2 were predisposed based on cavity volume, pocket lip size, hydrophobic volume, cavity surface area, and hydrogen-bond-forming surface area of the chosen HER2 cavity.
Figure 1a: Three-dimensional structure of HER2 and druggable cavities
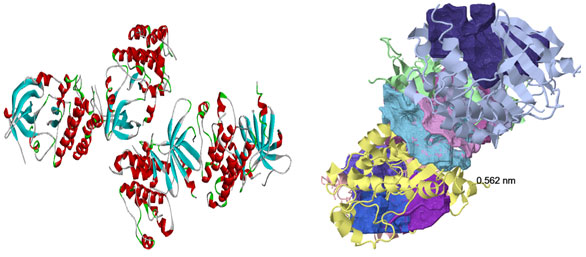
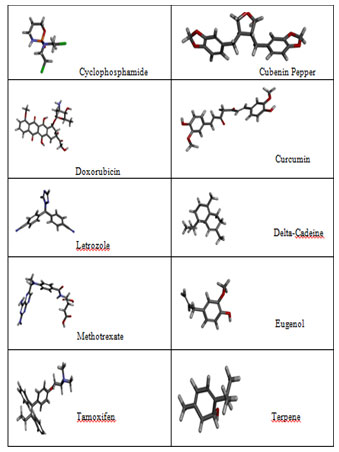
The Ligand Structures retrieved from Pubchem: PubChem an open source data repository for chemical substances and their related biological activities. This structure based search module compiled with variety of chemical structure format like SMILES, SMARTS, InChI, CID, molecular formula and SDF format. Since our ligandfit module accepts the SDF format we have downloaded all the Plant based natural compounds and synthetic compounds in SDF format. HER2 specific synthetic drugs as well natural compounds were listed in the Figure 2
Figure 2a: Natural compounds and synthetic compounds three dimensional structures
ADME Studies: ADME profiling of the natural compounds aids in proving the compounds are less toxic and it passes through various barriers.
Table 1. ADME Profiling of Natural Compounds
| Compound | ADMET-BBB | Solubility |
Hepatotoxicity Probability |
CYP2D6
Probability |
AlogP98 | ADMET_
PSA_2D |
|
| Cubenin | -0.203 | -4.448 | 0.708 | 0.227 | 3.194 | 65.466 | |
| Curcumin | -0.544 | -3.537 | 0.887 | 0.376 | 3.554 | 94.092 | |
| Delta Cadien | 1.373 | -5.713 | 0.258 | 0.178 | 4.939 | 0 | |
| Eugenol | 0.172 | -2.416 | 0.35 | 0.029 | 2.579 | 29.745 | |
| Terpene | 0.242 | -2.285 | 0.37 | 0.029 | 2.346 | 20.815 | |
Table 2. ADME Profiling of Synthetic Compounds
| Compound | ADMET-BBB | Solubility |
Hepatotoxicity Probability |
CYP2D6
Probability |
AlogP98 | ADMET_
PSA_2D |
|
| Cyclophosphamide | -0.723 | -1.296 | 0.589 | 0.059 | 0.33 | 42.393 | |
| Doxorubicin | -4.798 | 0.9 | 0.455 | -0.044 | 209.31 | ||
| Letrozole | -0.471 | -3.828 | 0.841 | 0.663 | 2.749 | 73.74 | |
| Methotrexate | -4.018 | 0.741 | 0.178 | 0.376 | 207.82 | ||
| Tamoxifen | 1.605 | -6.711 | 0.894 | 0.702 | 6.319 | 12.282 | |
Lipinski Rule : Lipinski rule of 5 helps in distinguishing between drug like and nondrug like molecules. It predicts high probability of success or failure due to drug likeness for molecules complying with 2 or more of the following rules:
Table 3. Lipinski Rule of Druggability for Synthetic Compounds
| MASS | HBD | HD A | Log P | MR | |
| Cyclophosphamide | 356 | 1 | 6 | 2.5102 | 91.47678 |
| Doxorubicin | 368 | 2 | 6 | 3.369898 | 102.0166 |
| Letrozole | 204 | 0 | 0 | 4.725199 | 66.74298 |
| Methotrexate | 164 | 1 | 2 | 2.1293 | 48.55979 |
| Tamoxifen | 152 | 1 | 1 | 2.2797 | 47.30179 |
Table 4. Lipinski Rule of Druggability for Natural Compounds
| MASS | HBD | HD A | Log P | MR | |
| Cubenin | 356 | 1 | 6 | 2.5102 | 91.47678 |
| Curcumin | 368 | 2 | 6 | 3.369898 | 102.0166 |
| Delta Cadeine | 204 | 0 | 0 | 4.725199 | 66.74298 |
| Eugenol | 164 | 1 | 2 | 2.1293 | 48.55979 |
| Terpene | 152 | 1 | 1 | 2.2797 | 47.30179 |
Molecular docking studies using ligandfit module : The docking studies where done to find the Receptor-Ligand Interactions LigandFit assess the small molecules affiinity into the HER2 protein active site by applyinng its shape complementary screening. In general docking process list the top conformations of ligand after the energy minimization method based on steepest descent method and conjugate gradient method. Ligandfit uses the consensus scoring to minimize the false positive. Proposed consensus scoring method was depends on LigScore1, LigScore2, piecewise linear potential 1 (PLP1) , piecewise linear potential 2, potential mean force (PMF) , Jain score.
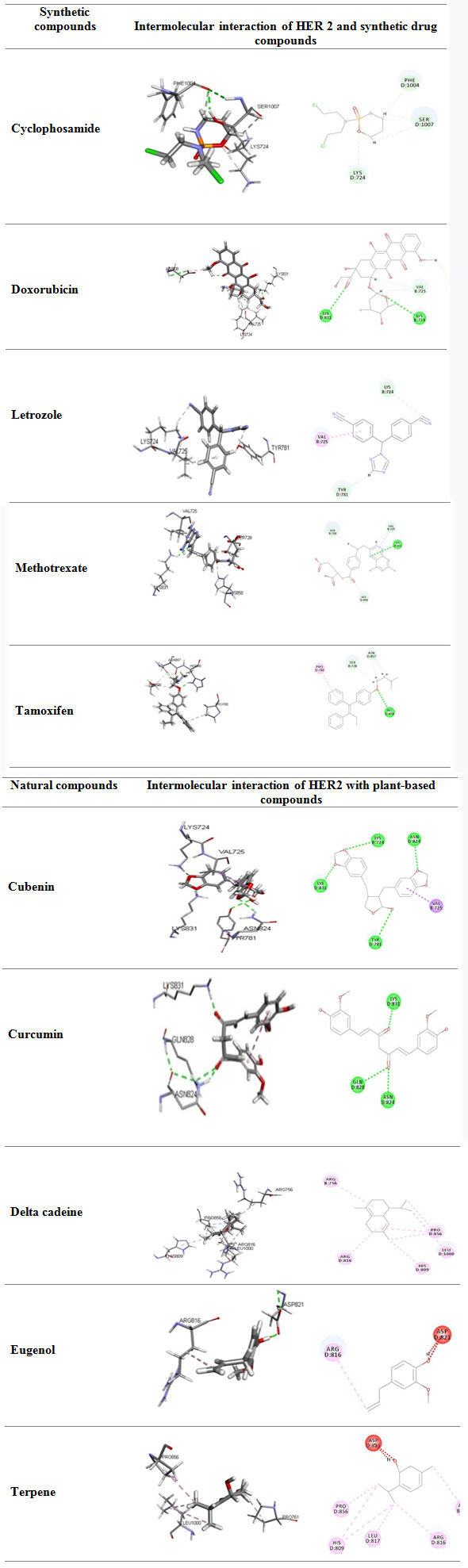
The above are the docking 2D & 3D images of the receptor with the ligand, with their docked positions and the respective amino acids. The dock scores are evaluated based on the parameters of lig score, PLP1, PLP2,JAIN score and PMF score.
Table 5. Dock scores of Synthetic compounds.
| Compounds | Lig score 1 | Lig score 2 | PLP1 | PLP 2 | JAIN | PMF | ||||||
| Cyclophosphamide | -0.168 | 1.652 | 30.288 | 32.14 | 1.035 | 13.01 | ||||||
| Doxorubin | 2.961 | 4.52 | 45.541 | 41.176 | 1.873 | 41.452 | ||||||
| Letrozole | 1.375 | 3.749 | 50.645 | 46.388 | -0.093 | 19.141 | ||||||
| Methotrexate | 2.883 | 4.105 | 38.079 | 30.442 | -2.452 | 34.579 | ||||||
| Tamoxifen | 1.878 | 3.238 | 34.473 | 32.632 | -1.709 | 41.811 | ||||||
Table 6. Dock scores of natural compounds
| Compounds | Lig score 1 | Lig score 2 | PLP1 | PLP 2 | JAIN | PMF |
| Cubenin | 3.563 | 4.065 | 47.011 | 48.088 | 0.271 | 37.173 |
| Curcumin | 3.235 | 4.174 | 45.657 | 44.984 | -1.815 | 50.533 |
| Delta Cadiene | 6.009 | 2.93 | 42.196 | 41.498 | 1.496 | 39.294 |
| Eugenol | 1.321 | 2.519 | 19.224 | 20.169 | 1.367 | 30.905 |
| Terpene | 1.368 | 2.549 | 26.964 | 29.576 | -0.044 | 33.962 |
CONCLUSION
Thus, the results of the docking studies carried out on HER2 corroborate to the findings that the most suitable drug like properties are possessed by the compound. In comparison with other compounds natural compounds are better and it is druggable. This provides evidence of how a natural compounds from plants can be a source of potential anti- cancer agent. The preclinical studies will pave way for a potential anti-cancer compound. The study aimed in finding the compatible natural lead molecules from plants and it shows that the compounds are druggable based on the Lipinski’s rule and the Insilico toxicity study studies are also positive, adding advantage for the compounds to be druggable. With the docking studies , the compounds that dock with the target has been found. The dock scores are also supporting for the further study. The work will be continued on breast cancer cell line study invitro and then in vivo studies.
REFERENCES
Aditi Gangopadhyay A Hirak Jyoti Chakraborty Abhijit Datta (2017) Targeting the Dengue β-OG With Serotype-Specific Alkaloid Virtual Leads Journal of molecular graphics & Modeling 73: 129. [PMID: 28279821]
Berman, HM J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne. (2000) The Protein Data Bank Nucleic Acids Research, 28: 235-242.
Cancer Genome Atlas Research Network, Weinstein, J. N., Collisson, E. A., Mills, G. B., Shaw, K. R., Ozenberger, B. A., Ellrott, K., Shmulevich, I., Sander, C., & Stuart, J. M. (2013). Nature genetics, 45(10), 1113–1120. https://doi.org/10.1038/ng.2764
Christy J & Swetha V (2019). Differential expression analysis of kruppel like factors 6 and antagonistic effect study of cinnamic acid – an in silico approach. Asian Journal of Pharmaceutical and Clinical Research, 12(7), 107-113. https://doi.org/10.22159 /ajpcr. 2019.v12i7.33406
Ishikawa T, Seto M, Banno H, et al.(2011) Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J Med Chem. 54(23):8030‐8050. doi:10.1021/jm2008634
Jain A.N.(1996) Scoring Noncovalent Protein-Ligand Interactions: A Continuous Differentiable Function Tuned to Compute Binding Affinities Journal of Computer-Aided Molecular Design,10,427–440
Jayaram, B Tanya Singh, Goutam Mukherjee, Abhinav Mathur, Shashank Shekhar, and Vandana Shekhar, Sanjeevini (2012) a freely accessible web-server for target directed lead molecule discovery BMC Bioinformatics 13, S7.
Kim, S., Thiessen, P. A., Bolton, E. E., Chen, J., Fu, G., Gindulyte, A., Han, L., He, J., He, S., Shoemaker, B. A., Wang, J., Yu, B., Zhang, J., & Bryant, S. H. (2016). PubChem Substance and Compound databases. Nucleic acids research, 44(D1), D1202–D1213. https://doi.org/10.1093/nar/gkv951
Krammer A, Kirchhoff PD, Jiang X, Venkatachalam CM, Waldman M.(2005) The Journal of Molecular Graphics and Modelling, 23,395-407
Nat. Rev. Mol. Cell Biol. 2,127–137
Pegram MD, Miles D, Tsui CK, Zong Y.(2020) Clin Cancer Res. 26(4):775‐786. doi:10.1158/1078-0432.CCR-18-1976
Siegel RL, Miller KD, Jemal A. CA (2020) Cancer J Clin. 70(1):7‐30. doi:10.3322/caac.21590
SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 18, 2714-2723.
Uhlen M. Cheng Zhang Sunjae Lee et al (2017). A pathology atlas of the human cancer transcriptome. Science. PubMed: 28818916 DOI: 10.1126/science.aan2507
Venkatachalam C.M., Jiang X., Oldfield T., Waldman M, (2003)The Journal of Molecular Graphics and Modelling,21, 289-07
Yarden, Y. & Sliwkowski,M X (2001) Kinase Inhibitor Drugs

