1, 3Department of Biotechnology, Mithibai College, Vile Parle (W), Mumbai 400056
2Department of Bioinformatics, Patkar-Varde College, Goregaon (W), Mumbai 400104
Corresponding author email: siddharth.sgr@gmail.com
Article Publishing History
Received: 18/04/2020
Accepted After Revision: 15/06/2020
Antibiotics are used for human therapy, farm animals and even in aquaculture. The overuse and misuse of these antibiotics result in pathogenic bacteria resistant to multiple drugs, which can be generated either by accumulating multiple genes on-resistance (R) plasmids or increased expression of multidrug efflux pump genes. The development of resistance in bacteria to conventional antibiotics restricts their use for treatment lead to the need for searching for an alternative to avoid this scenario. An antimicrobial protein of plant and animal origin is one of the most trending solutions for this issue. In our previous study, 7 such proteins were isolated from brain tissue lysate of Periplaneta americana. These proteins were further identified and characterized to be transferrin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using Mass Spectrometry techniques. The current study is aimed at analysing structure homology modeling and docking of transferring protein with its receptor on cell membrane of MRSA using the GRAMM-X docking server and further analysis using PDBsum. The results indicated significant interactions of Transferrin with its GAPDH receptor on MRSA. The results can be of future use to design an analogous drug based on the mechanism of action and binding site of transferrin on MRSA as well as probable ways of drug delivery for a better results.
Antibacterial protein, antibiotic resistance, mass spectrometry, protein docking
Sagar S, Javle V. R. K, Chunduri J. R. In Silico Modelling of Antibacterial Protein Transferrin from Periplaneta americana and its Interaction Analysis with Membrane Protein of MRSA. Biosc.Biotech.Res.Comm. 2020;13(2).
Sagar S, Javle V. R. K, Chunduri J. R. In Silico Modelling of Antibacterial Protein Transferrin from Periplaneta americana and its Interaction Analysis with Membrane Protein of MRSA. Biosc.Biotech.Res.Comm. 2020;13(2). Available from: https://bit.ly/2BfnYUp
Copyright © Sagar et al., This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) https://creativecommns.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
Bacterial infections and diseases are controlled by antibacterial drugs against pathogenic bacterial. The drug resistant bacteria ares one of the important causes for the death of about 50,000 new born annually. Insects are known to play an important role as carriers of these bacteria and well as spreading them amongst humans. The cockroach is found to harbor several gram-positive and gram-negative bacteria both internal and external of their digestive system (Sisai et al., 2006). In all around 114 multidrug-resistant strains were isolated from cockroach (Feleke et al., 2016). Almost all living organisms have innate immunity where they have the capability of developing antibacterial protein. Isolation of these proteins from different plant and animal sources has been trending these days (Zasloff, 2002). The 10 KDa protein isolated from brain tissue lysate of locust was found to be showing its antibacterial action against S.epidermis, S.auerus, MRSA and neuropathogenic E.coli K1. The antibacterial activity of the protein is heat resistant and is effective at a concentration as low as 5 μg. (Simon et al., 2012).
The polymyxins B and E (the latter is also known as colistin) are examples of antimicrobial peptides (AMPs) in clinical use since the 1950s. They have been applied for both topical and systemic treatment of infections (Landman et al., 2008). Their therapeutic use has increased to combat multidrug-resistant pathogens (Falagas and Kasiakou, 2005). However, their intravenous use has been limited by high nephrotoxicity and neurotoxicity (Falagas and Kasiakou, 2006). Identification of antibacterial proteins and their characterization is a very important aspect of proteomics nowadays. Without the identification of protein, its structure and its characteristics, it is difficult to reveal its action within the cell. Several techniques have been developed for the identification of the protein. Of them, Mass spectrometry has evolved as one of the most primary tools in protein identification (Thomson, 1913; Brunett et al,2020).
The use of mass spectrometry for biological applications dates from the 1950s (Beynon, 1965), and its use in peptide identification dates from the 1960s (Biemann et al., 1966). Accuracy, speed, and sample weight range have seen improvements spanning many orders of magnitude in recent decades (Henzel et al., 2003), making mass spectrometry one of the greatest scientific success stories of the twentieth century. For better extraction and identification of protein, mass spectrometry is combined with different other techniques like liquid chromatography (LC), gas chromatography (GC), time of flight (TOF), matrix-assisted laser desorption/ionization (MALDI), etc. (Jamer, 2009; Bilal et al., 2017; Francis et al., 2020).
Nano-capillary liquid chromatography -Electro Spray Ionization -Ion Trap -Time of Flight-Mass spectrometry/Mass Spectrometry (Nanocapillary LC–ESI–IT–TOF–MS/MS) has been successfully applied in clinical microbiology because of its economical and diagnostic benefits. It was used for rapid identification and comparison of S. aureus and its extracellular profile (Jones et al. 2008).
Most of the Antibacterial proteins show cationic property and using this they tend to destruct bacterial cell envelope. Antibacterial proteins target cell membranes through the formation of ion channels or trans-membrane pores and in this way destroy the bacterial cell (Duclohier, 2002; Park and Hahm, 2005). Apart from the membrane destruction, some AMPs, i.e. Pyrrhocoricin, Drosocin, and Apidaecin, may exert antibacterial activity by interactions with intracellular targets thus disrupting intracellular processes (Kragol et al., 2002; Li et al., 2006; Nicolas, 2009). The properties of protein can be better understood using in-silico tools at preliminary level and analyze their interactions with other proteins and those identified in/on drug resistant bacteria.
Interaction of antibacterial protein with the bacterial protein can be studied much better by identifying the structure of protein firstly. NMR or X-Ray crystallography are two standard techniques to identify the structure of the protein, however, it is time-consuming and expensive Gupta et al., 2014). Hence, the 3D structure prediction tool was used for the identification of the structure of nifA protein from Rhizobium leguminoserum (Sadam et al., 2018). To produce the tertiary structures of proteins, templates were selected from PDB (Protein Data Bank) (Bernstein et al., 1977; Goodsell et al, 2020) by using the BLASTp algorithm (Altschul et al., 1990; Lobanov et al,2020).
Sequences of proteins that are more similar to the query sequence, were selected as templates. The modeling of the three-dimensional structure of the proteins was performed by three homology modeling programs, Phrye2 (Kelley et al., 2015), SWISS MODEL (Arnold et al., 2006) and Modeller (Sali et al., 1993)
Bioinformatics tool gives great ease in predicting the protein-protein interaction using several algorithms which can later help in drug designing (Schmidt, 2013). Several Fast Fourier Transform – based docking programs have been made available as web servers e.g. ClusPro (Comeau et al., 2004), GRAMM-X (Tovchgrechko and Vakser , 2006) and ZDOCK (Chen et al., 2003). Overexpression of human epidermal growth factor receptor 2 (HER2) in the patients denotes poor prognosis leading to a reduced survival rate compared to other subtypes of breast cancer. Using GRAMM-X protein docking server interaction between Her2 receptor and protein from Pseudomonas exotoxin A (PE38) and A subunit of Shiga toxin 2a (Stx2a) was analyzed and gave promising results. (Goleij et al., 2019).
The analyses of docking are primarily image-based and include protein secondary structure, protein-ligand, and protein-DNA interactions, PROCHECK analyses of structural quality, and many others. This can be best achieved by the use of the PDBsum server (Roman et al., 2018). HAWKDOCK server uses Molecular Mechanics/Generalized Born Surface Area (MM/GBSA), which are more theoretically rigorous than scoring functions, have been widely used to predict binding free energies and identify correct binding conformations for protein-protein systems (Gaogi,2019). During the current study an attempt has been made to predict protein-protein interaction between transferrin and GAPDH as receptor on MRSA which can give us a lead for further research on its antibacterial pathway.
MATERIAL AND METHOD
Protein identification by TOF LC-MS/MS: LC-MS/MS was performed on an MS Q-TOF G65504A system. HPLC- Chip was used for ionization. After in-gel tryptic digestion peptides were eluted into the nano pump at the flow rate of 0.3 μL/min. Peptides were separated using the mobile phase gradient solvent A and Solvent B viz. Water and Acetonitrile respectively. Separation along mobile phase was in ratio of A 80% and B 20% for first 2 min, A 2% and B 98% for next 15 min, A 2% and B 98% for next 20 min, A 97% and B 3% for next 25min , A 97% B3% for next 35 min. The m/z ratio was between 300 – 3200 with an MS scan rate of 5.0 spectra/sec and MS/MS scan rate of 3.0 spectra/sec. LC-MS/MS data were acquired and protein was identified using NCBInr (https://www.ncbi.nlm.nih.gov/protein/) data.
Protein structure modelling: The structure of Protein identified using LC-TOF MS/MS was predicted using homology modeling server SWISS-MODEL (http://swissmodel.expasy.org/). The SWISS-MODEL server is based on homology-based modeling i.e. structure of the protein is predicted on sequence homology of the existing structure from the PDB database. SWISS-MODEL will search for templates initially form the PDB database, it allows selection of best template found and then generates the model with QMEAN4 score and Local quality estimate as evaluation parameter (Marco et al.,2014). The amino acid sequence of transferrin protein of cockroach and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) on the surface of the cell membrane of MRSA was extracted from the BLASTp server using accession no. 372292427 and KST21754.1 respectively. The FASTA sequence was entered and the server was allowed to form templates using its algorithms. Templates with maximum sequence identity and QMEAN value closest to 1 were selected for modeling the structure. The obtained structure prediction was downloaded in .pdb format and used for further docking.
Protein-Protein docking: GRAMM-X (http://vakser.bioinformatics.ku.edu/resources/gramm/grammx) server was used for online protein docking. GRAMM-X grew out of the original Fast Fourier Transformation (FFT) GRAMM methodology. It represents a new implementation that uses a smoothed Lennard-Jones potential on a fine grid during the global search FFT stage, followed by the refinement optimization in continuous coordinates and rescoring with several knowledge-based potential terms. The best surface match between molecules is determined by the correlation technique using FFT. An important feature of GRAMM is the ability to smooth the protein surface representation to account for possible conformational change upon binding within the rigid body docking approach (Andrey and Ilya, 2006).
The amino acid sequence of Transferrin was entered in ligand slot and an amino acid sequence of glyceraldehyde-3-phosphate dehydrogenase of MRSA cell membrane surface was entered in receptor slot. Visualization of different binding modeling was done using Discovery Studio (figure 2). HAWKDOCK server was used to identify the best binding pose for protein-protein in docking based on binding free energy score. The more the negative value more is the interaction and complex suitable to bind. The best model obtained was analyzed using a free online PDBsum server (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html). PDBsum-Generate module, at PDBSum allows the users to load protein (receptor) – protein (ligand) docked and analyze the structural characteristics and interactions are provided in simplified graphics. The analyses are primarily image-based and include protein secondary structure, protein-ligand, and protein-DNA interactions, PROCHECK analyses of structural quality (Roman et al., 2018).
The .pdb file obtained as result by GRAMM-X was uploaded on the PDBsum server. The link to the analytic result was obtained via mail. The functional annotation of the proteins was determined using the Universal Protein Resource (UniProt) database (http://www.uniprot.org/).
RESULTS AND DISCUSSION
Previously, around 7 antibacterial protein was isolated from brain tissue lysate of cockroach using Polyacrylamide Gel Electrophoresis (PAGE). These proteins were of molecular weight ranging from 97.4 kDa to 14.3 kDa. Amongst these, two proteins that showed the best antibacterial activity were chosen for its identification. TOF LC-MS/MS analysis revealed amino acid sequence and identified protein to be transferrin and glyceraldehyde-3-phosphate dehydrogenase having molecular weight 79.9 kDa and 35.5 kDa respectively (Table 1).
Table 1. Identification of protein using TOF LC-MS/MS analysis method
| Band no. | Num Spectra | Num Peps Unique | Score Unique | % Coverage | MW
(Da) |
pI | Species | Database | Accession no. | Entry name |
| 2 | 36 | 21 | 358.64 | 47.4 | 79914.4 | 5.43 | Periplaneta americana | NCBInr | 372292427 | Transferrin |
| 4 | 17 | 11 | 188.27 | 48.4 | 35585.1 | 6.98 | Periplaneta americana | NCBInr | 343965965 | Glyceraldehyde-3-phosphate dehydrogenase |
Protein structure modelling: The amino acid sequence and protein model of Transferrin of cockroach (figure 1a) and GAPDH of cell membrane surface of MRSA (figure 1b) was obtained using NCBInr and are as follows:
Transferrin

GAPDH at cell membrane of MRSA:
Protein-Protein Interaction: GRAMM-X online server showed multiple interactions of transferrin with three chains of GAPDH. Interactions were observed using PDBsum. It gave a good image (figure 3a) and picturesque prediction of interaction and bonds. Transferrin is denoted by letter E and chains of GAPDH is denoted by letter A, B, C, and D. It showed three types of interaction 1. Salt bridges are indicated by the red line, 2. Hydrogen bonds are indicated by blue line 3. Non bonded contacts are indicated by the orange line (figure 3b). Docking failed to show any disulfide interactions between proteins (generally indicated by the yellow line).
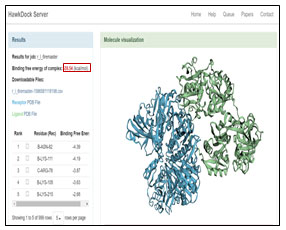
The GRAMM-X .pdb file showing the interaction between Transferrin and GAPDH was observed in Discovery Studio (figure 2). Out of 10 models of protein interaction that were obtained using GRAMM-X, model 6 was considered the best model due to lowest binding energy i.e -39.54 kcal/mol. The detail of same is given in figure 10 and table 3. EMBL-EBI’s PDBsum generated detailed reports of interaction. The following figure is a picturesque depiction of the interaction between transferrin which is denoted by Chain E and GAPDH consisting of four Chain A, Chain B, Chain C, and Chain D (figure 3b). Linear structure of protein is depicted in (figure 7).
Figure 1 : SWISS MODEL structure view in RasMol a. Transferrin, b. GAPDH of MRSA.
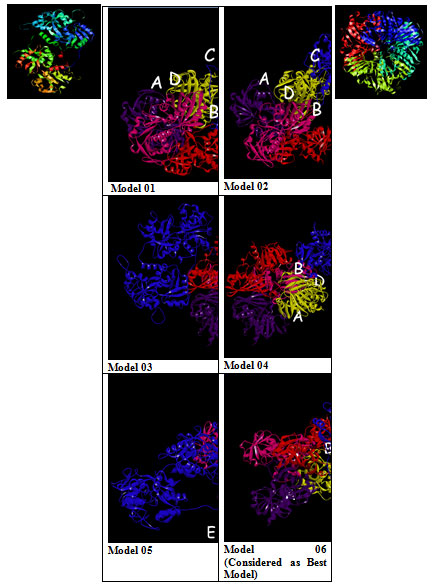
Figure 2: Model 1-10:- 3D View of protein interaction
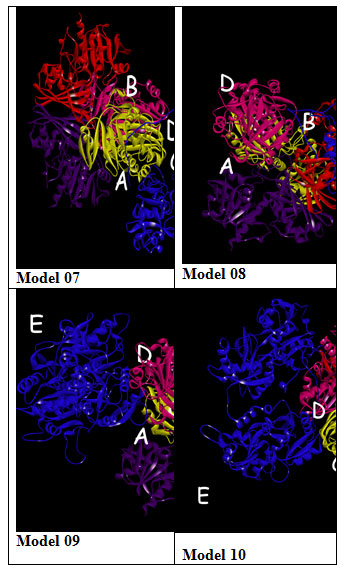
Figure 3: PDBsum report of GRAMM-X docking interaction of transferrin i.e E with chain A, B and C of GAPDH a. Cartoon model b. Picturesque of interaction.
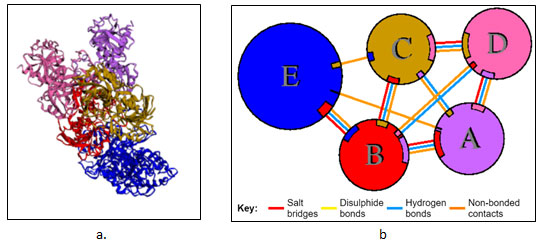
Table 2. List of types of interaction within the chain of GAPDH and between chains of GAPDH and transferrin.
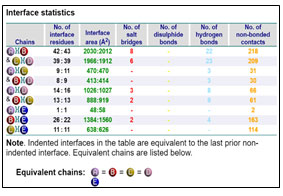
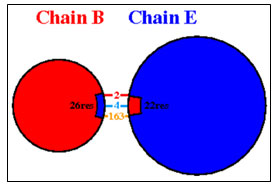
Interaction between Chain B of GAPDH with E
Chain B and Chain E share four hydrogen bonds, one between Asn82 and Thr513, Gln129 and Thr22, Glu143 and Ser311 and Asn136 and His24 each. There are 2 salt bridge, one between Lys111 and Glu509 and Glu143 and Arg307 each. Somewhere around 163 non-bonded contacts are observed. For these interactions, 26 amino acids of chain B and 22 amino acid residues of protein E are involved (figure 4 and figure 6). Figure 5 depicts cartoon model of interaction between Chain B of GAPDH and Chain E
Interaction between Chain C of GAPDH with E
Chain C and Chain E around 114 non-bonded contacts. For these interactions, 11 amino acids of chain C and 11 amino acid residues of protein E are involved (figure 2)
Interaction between Chain A of GAPDH with E
Chain A and Chain E share around 2 non-bonded contacts. For these interactions, 1 amino acids of chain A and 1 amino acid residues of protein E are involved (figure 2).
Figure 4: Picturesque Interaction between Chain E i.e transferrin and Chain B of GAPDH
(Above interaction may not be associated in one model but are given as combined from 10 different model)
Figure 5: Cartoon Image if interaction of Chain B GAPDH and Transferrin using PDBsum sever
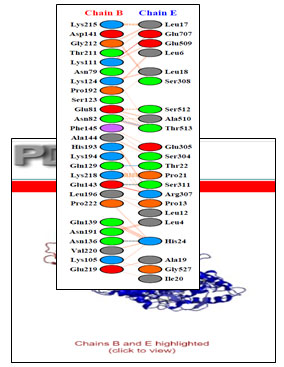
Figure 6: Detailed interactions of specific amino acids of Chain B of GAPDH and transferrin i.e Chain E

Figure 7: Secondary structure of Protein chain A, B, C, D and E
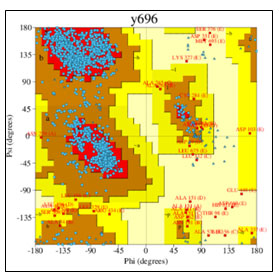
Figure 8: Ramachandran plot of detailed PROCHECK analyses
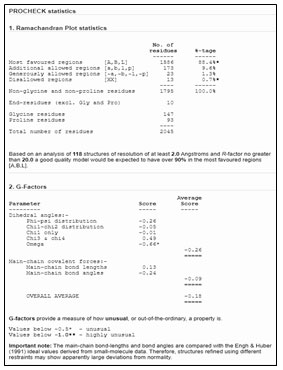
Figure 9: Detailed analysis of PROCHECK
Table 3: Calculating Binding Free energy for each complex generated from protein-protein docking
| Protein-Protein Docked complex from GRAMM-X Server | Binding Free energy Obtained from HAWKDOCK server (kcal/mol) |
| Complex 01 (r_l_1.pdb) | +09.20 |
| Complex 02 (r_l_2.pdb) | -04.28 |
| Complex 03 (r_l_3.pdb) | +00.59 |
| Complex 04 (r_l_4.pdb) | -01.65 |
| Complex 05 (r_l_5.pdb) | -04.36 |
| Complex 06 (r_l_6.pdb) | -39.54 |
| Complex 07 (r_l_7.pdb) | -06.91 |
| Complex 08 (r_l_8.pdb) | +10.63 |
| Complex 09 (r_l_9.pdb) | +08.76 |
| Complex 10 (r_l_10.pdb) | +15.62 |
Figure 10: Best Model Identified on Bases of Binding Free Energy Complex Using Hawkdock Server
The phenomenon of antimicrobial resistance has been known even before the introduction of penicillin as an antibiotic (Abraham and Chain, 1940) and over the last century, it has been observed that resistance arises independently from the chemical group of the drug. The issue has recently raised global awareness due to the increase of resistant organisms, the affected geographic locations and the breadth of resistance in single organisms (Levy and Marshall 2004; Chukwu et al.,2020).
Over the past twenty years, several studies have investigated the potential of cationic AMPs as novel anti-infective agents (Hancock and Lehrer, 1998; Marr et al., 2006). These naturally occurring compounds have been a model for the extensive design of new classes of antibacterial compounds (McGrath et al., 2013). Multiple such antibacterial proteins ranging from 64 kDa to 10 kDa have been isolated from cockroach and found to be effective against multiple drug-resistant and pathogenic bacteria, (Simon et al., 2012 Siddharth and Rao, 2015).
Insects produce these products and secrete in their hemolymph as their first line of defense mechanism (Seraj et al., 2003). Muga silkworm (Antheraea assamensis) larvae, when injected with Candida albicans; AMPs were isolated from the hemolymph and evaluated for antimicrobial activity against fungal and bacterial pathogens. Isolated peptides were confirmed by SDS-PAGE and TLC, and its molecular mass was determined as 9.052 kDa by MALDI-TOF mass spectrometry. From the mass fingerprinting analysis of this peptide after trypsin digestion, a peptide fragment with the molecular mass of 2622.7 Da was obtained. De novo sequencing of this peptide fragment following MS/MS analysis resembled with gloverin peptide of A. mylitta, comprising few aminoalkanoic acid residues as “KSGGGGWGS” with a complete score of 46.9. The peptide inhibited biofilm formation of the Gram-negative bacteria pathogens (Ellison et al., 1988). Iron-binding proteins like transferrin and lactoferrin express their antibacterial property by damaging the outer membrane of gram-negative and alter bacterial outer membrane permeability from an entry of iron (Waterhouse, 2018).
Net charge and amphipathic structure are the two interrelated features that confer antibacterial activity to AMPs. Often such peptides cause lysis of the bacterial cell and it is believed that cell lysis originates from the interaction between the bacterial anionic membranes and the positively charged peptides, (Skarnes and Watson, 1957; Raguse et al., 2002). Such interactions can best be predicted by using Bioinformatics tools. The X-ray structure of homodimer in chain A and B were used for autolysin while pneumolysin and pneumococcal surface protein A (pspA) was homology modeled with SWISS-MODEL where the peptides were based on NMR solution structure data of indolicidin peptide derivative (Le et al., 2015).Antimicrobial peptide interaction with the spike protein of the Coronavirus was checked using In silico tools Piper & GRAMM-X (Sabeena et al., 2019; Siddharth et al.,2020).
The mechanism of interaction between Death associated kinase 1 (DAPK1) and a subunit of antidepressant GluN2B-CT1290-1310 was predicted by GRAMM-X online server (Gao et al., 2018). GRAMM-X is the easy and reliable tool for prediction of protein-protein interaction (Pourjafar-Dehkordi et al., 2020). The stereochemical stability of the predicted models can be further verified using various protein quality based parameters such as percentage residues lying in favored and allowed regions, the number of glycine and proline residues and orientation of dihedral angles including phi (φ) and psi (ψ) and backbone conformation using PROCHECK module of the PDBSum server (Laskowski et al., 2005). Validation of docked complexes generated from GAMM-X suggested Model 6 had the best mode of binding with the free energy of -39.54 Kcal (Sarkar et al., 2020; Siddharth et al., 2020).
Majority of predicted residue were observed to be non-bonded interaction two for Chain A-E, 114 for Chain C-E. And residues from Chain B (ASN82, GLN129, GLY143, ASN136) shared four hydrogen bonding with Chain E (THR513, THR22, SER311, HIS24), also LYS111, GLU143(chain B) with GLU707, ARG307(chain E) formed two salt bridges that together with other interaction may act important from the perspective of stability of the complex and produce the required effect.
CONCLUSION
Transferrin protein that is produced in the brain tissue of cockroach showed an excellent antimicrobial capability against multiple bacteria both gram-negative and gram-positive. Glyceraldehyde-3-phosphate dehydrogenase found on the cell membrane of MRSA acts as a receptor for transferrin. This can be confirmed by using In silico tools like GRAMM-X which is a docking app helping us with providing information about protein interaction. Interaction can be understood in detail using EMBL-EBI’s PDBsum server.
ACKNOWLEDGEMENT
We would like to thank Dr. Rajpal Shripat Hande, Principal, SVKM’s Mithibai College for allowing us to use the infrastructure and facility of the Bioinformatics laboratory. We like to thank Ms. Manali Jadhav Co-ordinator of Mass Spectroscopy Department at SAIF-IIT Bombay for guiding and helping us obtain results of mass spectrometry.
REFERENCES
Abraham E.P., Chain E. (1940) An enzyme from bacteria able to destroy penicillin. Nature; 146(3713):837.
Altschul S.F. , Gish W. , Miller W. , Myers E.W. , Lipman D.J. (1990). Basic local alignment search tool J Mol Biol, 215 (3) , pp. 403-410
Arnold K. , Bordoli L. , Kopp J. , Schwede T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling Bioinformatics, 22 (2) , pp. 195-201
Bernstein, F. C., Koetzle, T. F., Williams, G. J., Meyer, E. F., Jr, Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T., & Tasumi, M. (1977). The Protein Data Bank: a computer-based archival file for macromolecular structures. Journal of molecular biology, 112(3), 535–542
Bertoni, M., Kiefer, F., Biasini, M., Bordoli, L., Schwede, T.(2017). Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Scientific Reports 7 .
Beynon, J.H. (1956). The use of the mass spectrometer for the identification of organic compounds. Microchimica Acta 44: 437.
Biemann K., Cone C., Webster B.R., Arsenault G.P. (1966). Determination of the amino acid sequence in oligopeptides by computer interpretation of their high-resolution mass spectra. J Am Chem Soc 88: 5598.
Bienert S., Waterhouse A., de Beer T.A.P., Tauriello G., Studer G., Bordoli L., Schwede T.(2017). The SWISS-MODEL Repository – new features and functionality. Nucleic Acids Res. 45, D313-D319
Bilal A., Madiha B., Muhammad A.N., Mohsin K., and Muhammad H.R. (2017), Proteomics: Technologies and Their Applications, Journal of Chromatographic Science, Vol. 55, No. 2, 182–196
Brunet, M.A., Leblanc, S. and Roucou, X., 2020. Reconsidering proteomic diversity with functional investigation of small ORFs and alternative ORFs. Experimental Cell Research, p.112057.
Chen R. , Li L. and Weng Z. (2003). ZDOCK: an initial-stage protein-docking algorithm. Proteins: Struct. Func. Bioinf., 52, 80–87.
Chukwu, E.E., Oladele, D.A., Awoderu, O.B., Afocha, E.E., Lawal, R.G., Abdus-salam, I., Ogunsola, F.T. and Audu, R.A., 2020. A national survey of public awareness of antimicrobial resistance in Nigeria. Antimicrobial Resistance & Infection Control, 9, pp.1-10.
Comeau,S.R., Gatchell,D.W., Vajda,S. and Camacho,C.J. (2004). ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res., 32, W96–W99.
Duclohier H., (2002). How do channel- and pore-forming helical peptides interact with lipid membranes and how does this account for their antimicrobial activity? Mini-Rev. Med. Chem. 2, 331–342.
Ellison R.T., Giehl T.J., LaForce F.M., (1988). Damage of the outer membrane of enteric gram-negative bacteria by lactoferrin and transferrin, Infect Immun.; 56(11):2774-81.
Falagas M.E., Kasiakou S.K.(2006), Toxicity of polymyxins: a systematic review of the evidence from old and recent studies. Crit Care 2006; 10(1):1
Falagas M.E., Kasiakou S.K.(2005). Colistin: the revival of polymyxins for the management of multidrug resistant gram-negative bacterial infections. Clin Infect Dis; 40:1333-1341.
Feleke M., Setegn E., Mengistu E., Kahsay H., Dagnachew M., Tigist F., Fisha G., Getenet A., and Raja N. (2016). Cockroaches as a Source of High Bacterial Pathogens with Multidrug Resistant Strains in Gondar Town, Ethiopia, BioMed Research International, Volume 2016, Article ID 2825056,pg.no. 1-6
Gao T. ,Tingting F. ,Fengyuan Y. ,Lixia Y. ,Weiwei X. ,Feng Z. (2018). Prediction of GluN2B-CT1290-1310/DAPK1 Interaction by Protein–Peptide Docking and Molecular Dynamics Simulation, Molecules , 23(11), 3018
Francis, F., Mazzucchelli, G., Baiwir, D., Debode, F., Berben, G. and Megido, R.C., 2020. Proteomics based approach for edible insect fingerprinting in novel food: Differential efficiency according to selected model species. Food Control, 112, p.107135.
Gaoqi W., Ercheng W., Zhe W., Hui L., Feng Z., Dan L. and Tingjun H 2019). HawkDock: a web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA W322–W330 Nucleic Acids Research, Vol. 47
Goleij Z. , Mahmoodzadeh H. H. , Amin M. , Amani J. , Behzadi E., Abbas A. I.F. (2019). In Silico Evaluation of Two Targeted Chimeric Proteins Based on Bacterial Toxins for Breast Cancer Therapy, Int J Cancer Manag.; 12(2):e83315
Goodsell, D.S., Zardecki, C., Di Costanzo, L., and Young, J.Y., (2020). RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Science, 29(1), pp.52-65.
Guex, N., Peitsch, M.C., Schwede, T.(2009). Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 30, S162-S173 .
Gupta C.L., Akhtar S. , Bajpai P. (2014), In silico protein modeling: possibilities and limitations EXCLI J., 13 , pp. 513-515
Hancock R.E.W., Lehrer R.(1998) Cationic peptides: a new source of antibiotics. Trends Biotechnol;16:82-88.
Henzel W.J., Watanabe C., Stults J.T. (2003). Protein identification: The origins of peptide mass fingerprinting. J Am Soc Mass Spectrom 14: 931–942
James J.P.(2009). Principles and Applications of Liquid Chromatography- Mass Spectrometry in Clinical Biochemistry, Clin Biochem Rev Vol 30. 19-34
Jones R.C., Deck J., Edmondson R.D., Hart M.E. (2008). Relative quantitative comparisons of the extracellular protein profiles of Staphylococcus aureus UAMS-1 and its sarA, agr, and sarA agr regulatory mutants using one-dimensional polyacrylamide gel electrophoresis and nanocapillary liquid chromatography coupled with tandem mass spectrometry. J Bacteriol 190(15): 5265–5278
Kelley A. , Mezulis S. , Yates C.M. , Wass N.M. , Sternberg M.J.E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis Nature Protocols, 10 , pp. 845-858
Kragol G., Hoffmann R., Chattergoon M.A., Lovas S., Cudic M., Bulet P., Condie B.A., Rosengren K.J., Montaner L.J., Otvos L. Jr, (2002). Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin. Eur. J. Biochem. 269, 4226–4237
Landman D., Georgescu C., Martin D.A., Quale J. (2008). Polymyxins revisited. Clin Microbiol Rev: 21: 449-465.
Laskowski, R. A., Chistyakov, V. V., and Thornton, J. M. (2005). PDBSum more: new summaries and analysis of the known 3D structure of proteins and nucleic acids. Nucleic Acids Res. 33, 266–268.
Le C.F., Yusof M.Y., Hassan M.A., Lee V.S., Isa D.M., Sekaran S.D.(2015). In vivo efficacy and molecular docking of designed peptide that exhibits potent antipneumococcal activity and synergises in combination with penicillin. Sci Rep. 2015;5:11886.
Levy S.B., Marshall B. (2004). Antibacterial resistance worldwide: causes, challenges and responses. Nat Med; 10:S122-S129.
Li W.F., Ma G.X., Zhou X.X., (2006). Apidaecin-type peptides: Biodiversity, structure–function relationships and mode of action. Peptides 27, 2350–2359
Lobanov, M.Y., Likhachev, I.V. and Galzitskaya, O.V., 2020. Disordered Residues and Patterns in the Protein Data Bank. Molecules, 25(7), p.1522.
Marco B., Stefan B., Andrew W., Konstantin A., Gabriel S., Tobias S., Florian K., Tiziano G.C., Martino B., Lorenza B.,Torsten S.,(2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information, Nucleic Acids Research, Vol. 42, W252–W258.
Marr A.K., Gooderham W.J., Hancock R.E.W(2006). Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol; 6:468-472.
McGrath D.M., Barbu E.M., Driessen W.H., Lasco T.M., Tarrand J.J., Okhuysen P.C., Arap W.(2013). Mechanism of action and initial evaluation of a membrane active all-D-enantiomer antimicrobial peptidomimetic. Proc Natl Acad Sci USA; 110(9):3477-3482.
Nicolas P., (2009). Multifunctional host defence peptides: intracellular-targeting antimicrobial peptides. FEBS J. 276, 6483–6496
Park Y., Hahm K.S., (2005). Antimicrobial peptides (AMPs): peptide structure and mode of action. J. Biochem. Mol. Biol. 38, 507–516.
Pourjafar-Dehkordi, D. and Zacharias, M., 2020. Prediction of protein–protein complex structures by docking. Protein Interactions: Computational Methods, Analysis And Applications, p.59.
Raguse T.L., Porter E.A., Weisblum B., Gellman S.H.(2002). Structure-activity studies of 14-helical antimicrobial beta-peptides: probing the relationship between conformational stability and antimicrobial potency. J Am Chem Soc 2002; 124(43):12774-12785.
Roman A. L. , Jagoda J., Lukas P., Radka S.V., and Janet M. T. (2018). PDBsum: Structural summaries of PDB entries, Protein Science vol 27:129—134
Sabeena M., Hanan B., and Musa G. (2019). Peptide-Protein Interaction Studies of Antimicrobial Peptides Targeting Middle East Respiratory Syndrome Coronavirus Spike Protein: An In Silico Approach, Advances in Bioinformatics Volume 2019, Article ID 6815105, 1-16
Sadam D.V. S, Krishna M.S.R., Pindi P.K., Sirisha J.,(2018). In silico structural homology modeling of nif A protein of rhizobial strains in selective legume plants, Journal of Genetic Engineering and Biotechnology, Volume 16, Issue 2, Pages 731-737
Sali A., Blundell T.L. (1993). Blundell Comparative protein modelling by satisfaction of spatial restraints J Mol Biol, 234 (3), pp. 779-815
Sarkar, B. and Ullah, M.A., 2020. Designing Novel Subunit Vaccines against Herpes Simplex Virus-1 using Reverse Vaccinology Approach. bioRxiv.
Schmidt T., Bergner A. and Schwede T., (2013). Modelling three-dimensional protein structures for applications in drug design. Drug Discov. Today, 2014 Jul;19(7):890-7
Seraj U.M. , Hoq M.I. , Anwar M.N. and Chowdhury S. (2003), A 61kDa antibacterial protein isolated and purified from the hemolymph of the American cockroach Periplaneta amricana, Pak J Biol. Sci., vol.6 pp.715-720.
Siddharth S. and JayaPrada Rao C. (2015), Periplaneta species brain proteins and their efficacy as antibiotics, Annual International Conference on Advances in Biotechnology (Bio Tech-2015) Edition: March.Pg. No. 102-105
Siddharth S, Jayaprada R C and Vyomesh J (2020); An Assessment Of The Interaction Between Insect Brain Protein And Non-Structural Protein Of Coronavirus Using In-Silico Analyses Int. J. of Adv. Res. 8 (May). 436-452
Simon L., Naveed A.K., Ruqaiyyah S. (2012),.Animals living in polluted environments are potential source of antimicrobials against infectious agents, Pathogens and Global Health, volume 106 -4 , pg.218-223.
Sisai M., Ignatius M. M., Berhanu A. G., Joseph A., Gervus M. and Neema M. (2006). Microbiological studies of cockroaches from three localities in Gaborone, Botswana. Aftican Journal of food agriculture nutrition and development.6 (2):1-17.
Skarnes R.C., Watson D.W.(1957). Antimicrobial factors of normal tissues and fluids. Bacteriol Rev;21:273-294
Studer, G., Rempfer, C., Waterhouse, A.M., Gumienny, G., Haas, J., Schwede, T.(2020). QMEANDisCo – distance constraints applied on model quality estimation. Bioinformatics 36, 1765-1771.
Thomson J.J. (1913). Rays of positive electricity and their application to chemical analysis. Proc Roy Soc 89: 1–20.
Tovchigrechko A. and Vakser I.A. (2006). GRAMM-X public web server for protein-protein docking. Nucleic Acids Res., 34, W310–W314.
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F.T., de Beer, T.A.P., Rempfer, C., Bordoli, L., Lepore, R., Schwede, T (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296-W303
Zasloff M. (2002). Antimicrobial peptides of multicellular organisms. Nature;415:389, 95.

