G. Rajitha,1*![]() M. VidyaRani,1
M. VidyaRani,1![]() V. Umakanth Naik2
V. Umakanth Naik2![]() and A. Umamaheswari2
and A. Umamaheswari2![]()
1Institute of Pharmaceutical Technology, Sri Padmavati Mahila Visvavidyalayam, Tirupati, India
2Department of Bioinformatics, Sri Venkateswara Institute of Medical Sciences, Tirupati,India
Corresponding author email: rajitha.galla@gmail.com
Article Publishing History
Received: 18/11/2021
Accepted After Revision: 25/03/2022
EGFR (Epidermal Growth factor receptors) expressed in different type of cancers, such as breast, esophageal, lung cancer etc. Because of their multifaceted role in cancer progression, EGFR and its related receptors have been considered as attractive targets for developing anticancer agents, i.e. EGFR inhibitors consisting molecules targeting EGFR ATP binding pocket and monoclonal antibodies targeting EGFR ligand binding domain. For patients with EGFR-mutant non-small-cell lung cancer, acquired resistance to drugsobstructs long-term therapeutic efficacy of targeted therapy. Even though third-generation medicines targeting EGFR T790M mutation have shown promise in overcoming acquired resistance to EGFR-tyrosine kinase inhibitors, fourth-generation drugs targeting acquired resistance to 3rdgeneration inhibitors are still needed.Hence, in present study, EGFR (PDB: 1M17) was selected as a target to perform molecular docking studies for existing ligands from literature. Interaction fingerprint analysis was applied for dockedcomplexes. Docking studies revealed that compound 3exhibited good binding affinity towards the selected target with XPG score-9.439Kcal/Mol among existing ligands which is comparable with that of standard drug erlotinib -9.192 Kcal/Mol. Interaction fingerprint analysis further confirmed that best docked compounds showed H-bond interaction with backbone residue of Met 769 and Cys 773 in an identical manner with that of standard drug erlotinib.Present study concludes thatamong selected existing compounds, the ligands containing quinazoline nucleus exhibited good binding affinity and similar binding interactions when compared with that of standard drug erlotinib and these ligandscan be further optimized to increase binding affinity and interactions with the selected target.
EGFR, heterocyclic, interaction finger print, Docking.
Rajitha G, Rani M. V, Naik V. U, Umamaheswari A. Design of Heterocyclic Compounds as Epidermal Growth Factor Receptor Inhibitors using Molecular Docking and Interaction Fingerprint Studies. Biosc.Biotech.Res.Comm. 2022;15(1).
Rajitha G, Rani M. V, Naik V. U, Umamaheswari A. Design of Heterocyclic Compounds as Epidermal Growth Factor Receptor
Inhibitors using Molecular Docking and Interaction Fingerprint Studies. Biosc.Biotech.Res.Comm. 2022;15(1). Available from: <a href=”https://bit.ly/3KlqsP0“>https://bit.ly/3KlqsP0</a>
Copyright © This is an Open Access Article distributed under the Terms of the Creative Commons Attribution License (CC-BY). https://creativecommons.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provided the original author and sources are credited.
INTRODUCTION
Cancer is the major cause of death worldwide, accounting for nearly 10 million deaths in 2020 (Sung et al. 2021). EGFR (Epidermal Growth factor receptor) is a major factor in epithelial malignancies, and EGFR activity increases tumor growth, invasion and tumor metastasis. In normal tissues, the EGFR ligands availability is strictly controlled to make sure that the cell proliferation kinetics absolutely meet the requirements of tissues to maintain homeostasis. In the tumor microenvironment, EGFR is constantly stimulated because of continuous production of EGFR ligands or as a result of EGFR mutation that sticks receptor in a continuous activation state. Hence, EGFR has been considered as an important target for anticancer agents(Sasaki et al. 2013;Tsubata 2021).
Attempts were made to the structural modification of heterocyclic compounds to discover powerful anticancer agents. Pyrazole & fused pyrazoles, viz., pyrazolpyrimidine derivatives and pyranopyrazolesare promising scaffolds for numerous anticancer agents(Saleh et al. 2020). Pyrimidine based, third generation irreversible EGFR inhibitors are WZ4002 and osimertinib (Engel et al. 2015; Greig et al. 2016;Wu et al. 2020). Various thiophene based derivatives were reported as dual EGFR or HER2 (human EGFR-related receptor 2) inhibitors (Xiao et al. 2020).
Quinazoline-based EGFR tyrosine kinase inhibitors such aserlotinib, lapatinib etc.are available in the market for treatment of nearly 30% of Non-small cell lung carcinoma cases(Bhatia et al. 2020). Hence, in our present study, existing ligands containing heterocyclic compounds were retrieved from literature for molecular docking studies against EGFR to determine the binding affinity and interactions of existing ligands containing different nucleuswith the selected target (Tsubata 2021).
MATERIAL AND METHODS
Existing EGFR inhibitors with heterocyclic nucleus were retrieved from literature (Li et al. 2011; Wang et al. 2016; Tu et al. 2016; Toolabi et al. 2019). Structure of co-crystal ligand AQ4 (Erlotinib) was collected from PDB database. Ligands were prepared using LigPrep application with SchrödingerEpik module, such asenhancement of stereochemical nature and protonation state of ligand, development of tautomers and minimization of energy using force field OPLS-3 at pH7.0±2.0 (Friesner et al. 2004;Pradeep et al. 2015).The best resolute X-ray structure of EGFR tyrosine kinase domain with 4-anilinoquinazoline inhibitor erlotinib (1M17) was selected in present study to propose EGFR antagonists by molecular docking & protein-ligand interaction fingerprint analysis (Jennifer et al. 2002).
Literature report indicated that 4-anilinoquinazolines like erlotinib inhibits EGFR by binding to site which is occupied by ATP during phosphotransfer. EGFR crystal structure was imported to Maestro and protein structure was prepared using protein preparation wizard. The optimization of H-bonding network was done by reorientation of hydroxyl groups and thiol groups in protein; other operations were performed which were not part of refinement process of X-ray crystal structure(Sudheer et al. 2021).
Protein optimization was done at neutral pH and then minimization of energy was done by applying OPLS-3 force field for all atoms. Grid was generated around target key residues using Glide v7.1. Removal of the undesirable water molecules was done using Protein preparation wizard from the inhibitor binding site of target (Sudheer et al.2021).GLIDE XP (grid-based ligand docking with energetic extra precision) docking procedure was used for determining target & ligandbinding affinity (Duch et al. 2007). The prepared ligands were dockedflexibly into grid box which was generated around the inhibitor binding site residues of EGFR using Monte Carlo-based simulated algorithm minimization method (Sudheer et al. 2021). Binding, binding orientation and ranking were represented by Glide Score. During XP docking,ten poses were generated for each ligand and after post-docking minimization the best pose was retained.
Selected existing ligands were subjected to QikProp module of Schrödinger suite for prediction of the pharmacological descriptors and ADME properties. In addition, SASA and related values of docked ligands were also determined using Schrödinger suite (Thirupathi et al. 2016).Docking interactions were further analyzed by interaction fingerprint studies to determine whether the test compounds showed similar interactions like standard drug. Interaction fingerprint was generated for the best docked compounds and erlotinib docked complexes (Deng et al. 2004). Value 1 was given for established interactionsand 0 for absence of specific interaction (Rácz et al. 2018).
RESULTS AND DISCUSSION
The structures of 77 EGFR inhibitors were collected from literature, and erlotinib structure was retrieved from PDB database and prepared using LigPrep application withSchrödinger Epik module.The PDB id 1M17 was selected to determine the antagonist behavior of existing ligands. The protein preparation wizard was used for minimizing energies of prepared crystal structure of EGFR. Inhibitor binding site residues were defined around the grid generated within the EGFR. The EGFR-AQ4 (Erlotinb) complex inhibitor binding site comprises residues such as Leu 694, Gly 695, Ser 696, Ala 719, Lys 721, Glu 738, Leu 764, Thr 766, Glu 767, Leu 768, Met 769, Pro 770, Phe 771, Gly 772, Cys 773, Leu 820, Thr 830, Asp 831, Phe 832 within the 4 Å region surrounding AQ4. The binding site residues of EGFR were defined 4 Å around co-crystal ligand using PDBsum.
Table 1. Docking scores of EGFR with erlotinib and best docked existing compounds
| S.No | Compound code | XPG Score (Kcal/Mol) |
| 1. | Erlotinib | -9.192 |
| 2. | 3 | -9.439 |
| 3. | 7 | -9.402 |
| 4. | 24 | -8.863 |
| 5. | 33 | -8.291 |
About 77 existing ligands from literature and Erlotinib were docked with EGFR. Among all the docked complexes, Compound 3exhibited least XPGscore of -9.439 Kcal/mol with good interactions with key amino acid residues (Table 1).Pharmacological descriptors and ADME/T profile of erlotinib and best docked ligands were determined using epic module of schrodinger.Results indicated that the selected existing ligands showed favorable ADME/T properties within the range of 95% FDA approved drugs. Results were represented in table 2 & table 3(Pradhan et al.2013).
Table 2. Pharmacological descriptors of erlotinib and best docked compounds
| Comp. Code | M.W | SASA | WPSA | FOSA | PISA | Vol | HBD | HBA | Glob | IP |
| Erlotinib | 393.441 | 714.826 | 0 | 374.94 | 279.021 | 1278.941 | 1.5 | 7.4 | 0.797135 | 8.396 |
| 3 | 609.451 | 856.13 | 114.902 | 338.114 | 311.952 | 1620.635 | 2 | 8.95 | 0.779379 | 8.382 |
| 7 | 594.396 | 848.926 | 114.901 | 188.262 | 356.093 | 1549.14 | 2 | 8.45 | 0.762703 | 8.591 |
| 24 | 574.408 | 852.538 | 114.901 | 183.498 | 385.154 | 1545.105 | 2 | 8.95 | 0.758153 | 8.542 |
| 33 | 424.879 | 597.812 | 111.28 | 46.685 | 337.15 | 1138.137 | 2 | 5 | 0.881855 | 8.72 |
MW = Molecular weight(130/725); SASA = Total solvent accessible surface area(300/1000); WPSA= Weakly polar solvent accessible surface area(0/175); FOSA = Hydrophobic solvent accessible surface area(0/750); PISA = Carbon Pi solvent accessible surface area (0/450); Vol = Molecular volume (A^3)(500/2000); HBD = Hydrogen bond Donor (0/6); HBA = Hydrogen bond acceptor(2/20); Glob = Globularity(0.75/0.95); IP (eV) = Ionization potential(7.9/10.5)
Table 3. ADME/T properties of erlotinib and best docked compounds
| Compounds | Rule of 5 | Log P o/w | Log BB | Log KhSa | Log KP | Rule of 3 |
| Erlotinib | 0 | 4.053 | -0.738 | 0.157 | -0.602 | 0 |
| 3 | 2 | 5.738 | -0.758 | 0.776 | -1.141 | 1 |
| 7 | 1 | 4.854 | -1.871 | 0.714 | -2.897 | 1 |
| 24 | 1 | 4.795 | -1.657 | 0.602 | -2.413 | 1 |
| 33 | 0 | 4.319 | -0.405 | 0.473 | -1.648 | 0 |
Rule of 5: Violations of Lipinski’s Rule of 5 (Max 4); Log P o/w = log P octanol/water (1.80/6.50); Log BB = log BB brain/blood (-3/1.2); Log KhSa = serum protein binding (-02.5/01.5); Log KP = skin permeability (KP in cm/h); Rule of 3: Violations Jorgensen Rule of 3 (Max 3)
The co-crystal ligand erlotinb exhibited two hydrogen bond interactions with back bone residues of Met 769 and Cys 773 respectively. Among all the selected heterocyclic compounds, the compounds containing quinazoline heterocyclic nucleus (compounds3, 7 and 24) exhibited good interactions with key aminoacid residues and two H-bonds with backbone residues of Met 769 and Cys 773 with docking scores -9.439Kcal/Mol, -9.402Kcal/Mol and -8.863Kcal/Mol respectively which is comparable to that of standard drug erlotinib. In addition to the interactions with Met 769 and Cys 773, compound 7 showed one H-bond interaction with backbone residue of Ser 696 indicating good EGFR inhibition.
Among the compounds containing thienopyrimidine nucleus, compound 33 showed moderate binding affinity with XPG score -8.291 Kcal/Mol and exhibited only one H-bond interaction with Met 769 (Fig 1-5). All the best docked compounds exhibited good interaction with “hing region key residue” Met769 of EGFR, which is involved in the anticancer treatment strategies.Best docked compounds 3, 7 and 24 exhibited H-bond interaction with backbone residues of key amino acid Cys-773 which is positioned within the EGFR ATP binding pocket(Fry et al. 1998; Nasab et al. 2018; Sudheer et al. 2021).
Figure 1: Interactions of erlotinib with EGFR
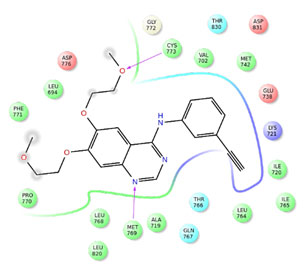
Figure 2: Interactions of compound 3 with EGFR
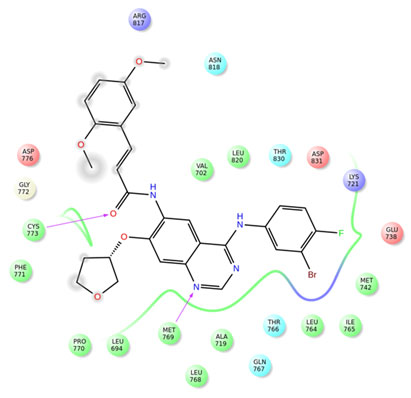
Figure 3: Interactions of compound 7 with EGFR
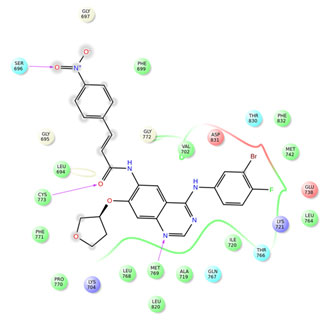
Figure 4: Interactions of compound 24 with EGFR
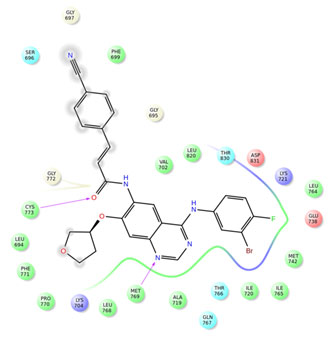
Figure 5: Interactions of compound 33 with EGFR
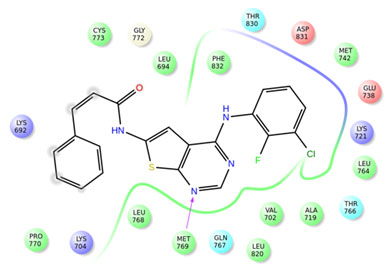
Table 4: Interaction fingerprint of the erlotinib and best docked compounds
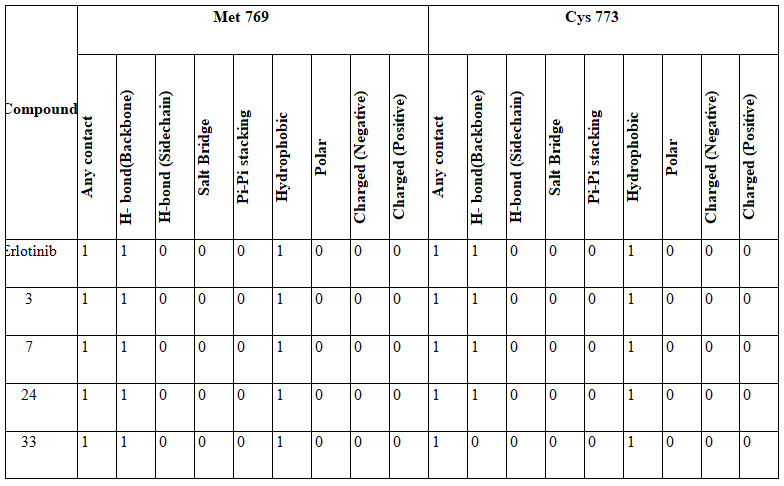
A 9bit interaction fingerprint was generated to determine interaction of best docked compounds in the inhibitor binding site of hERα(Table 4) and compared with standard drug erlotinib. Results indicated that compounds with quinazoline nucleus exhibited similar interactions with that of erlotinib.
CONCLUSION
The findings of present study can help in designing the novel EGFR inhibitors with better binding affinity and interactions by using molecular docking studies of existing EGFR inhibitors against EGFR (1M17) and interaction fingerprint analysis. Among all the selected existing ligands, compound 3 exhibited good binding affinity with XPG score -9.439 Kcal/Mol which is comparable with that of standard drug erlotinib. Interaction fingerprint analysis further confirmed that the best docked compound showed two H-bond interactions with backnone residues of Met 769 and Cys 773 similar to that of standard Erlotinib. Results of the present study concludes that these derivatives properly fit into the ATP binding pocket in EGFR crystal structure with good binding affinity, indicating that they may act as potential EGFR inhibitors.
ACKNOWLEDGEMENTS
The study is financially supported by the CURIE-AI Center, SPMVV under the “Innovative Research projects to the faculty (Minor Projects) to develop AI Databases” of CURIE- AI Phase-II Recurring grant.
Conflict of Interests: Authors declare no conflicts of interests to disclose.
Funding: The study is financially supported by the CURIE-AI Center, SPMVV under the “Innovative Research projects to the faculty (Minor Projects) to develop AI Databases” of CURIE- AI Phase-II Recurring grant.
Data Availability Statement: The database generated and /or analysed during the current study are not publicly available due to privacy, but are available from the corresponding author on reasonable request.
REFERENCES
Bhatia, P, Sharma, V, Alam, O, et al.(2020). Novel quinazoline-based EGFR kinase inhibitors: A review focussing on SAR and molecular docking studies (2015-2019),European Journal of Medicinal Chemistry, vol. 204, pp. 112640.
Dasari, T,Kondagari, B, Dulapalli, R,et al.(2016). Design of novel lead molecules against RhoG protein as cancer target – a computational study,Journal of Biomolecular Structure and Dynamics,vol. 35, no. 14, pp. 3119–3139.
Deng, Z, Chuaqui, C and Singh, J (2004). Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein-Ligand Binding Interactions,Journal of Medicinal Chemistry, vol. 47, no. 2, pp. 337-344.
Duch, W, Swaminathan, K and Meller, J (2007).Artificial Intelligence Approaches for Rational Drug Design and Discovery,Current Pharmaceutical Design,vol. 13, no. 14, pp. 1497–1508.
Engel, J, Richters, A, Getlik, M, et al.(2015).Targeting Drug Resistance in EGFR with Covalent Inhibitors: A Structure-Based Design Approach,Journal of Medicinal Chemistry, vol. 58, no. 17,pp. 6844–6863.
Friesner, RA, Banks, JL, Murphy, RB,et al.(2004). Glide: A new approach for rapid, accurate docking and scoring: Method and assessment of docking accuracy,Journal of Medicinal Chemistry,vol. 47, no. 7, pp. 1739–1749.
Fry, DW, Bridges, AJand Denny, WA (1998).Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor, Proceedings of the National Academy of Sciences of the United States of America,vol. 95, no. 20, pp. 12022-12027.
Greig, SL2016,‘Osimertinib: First Global Approval,Drugs,vol. 76, no. 2, pp. 263-73.
Jennifer, S, Sliwkowski, MX andEigenbrot, C (2002). Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor,Journal of Biological Chemistry,vol. 277, no. 48, pp. 46265-46272.
Li, DD, Lv, PC, Zhang, HJ,et al. (2011). The combination of 4-aminoquinazoline and cinnamic acid: A novel mode of binding to the epidermal growth factor receptor tyrosine kinase,Bioorganic & Medicinal Chemistry,vol. 19, no. 16, pp. 5012-5022.
Nasab, RR, Mansourian, M, Hassanzadeh, F,et al.(2018). Exploring the interaction between epidermal growth factor receptor tyrosine kinase and some of the synthesized inhibitors using combination of in-silico and in-vitro cytotoxicity methods,Research in Pharmaceutical Sciences,vol. 13, pp. 509-522.
Pradeep, N, Munikumar, M, Swargam, S, et al. (2015). 197 Combinations of e-pharmacophore modeling, multiple docking strategies and molecular dynamic simulations to discover of novel antagonists of BACE1,Journal of Biomolecular Structure and Dynamics,vol. 33, no. 1, pp. 129-130.
Rácz, A, Bajusz, D and Héberger, K (2018). Life beyond the Tanimoto coefficient: similarity measures for interaction fingerprints,Journal of Cheminformatics,vol. 10, pp. 1-12.
Saleh, NM, El-Gazzar, MG, Aly, HM, et al.(2020). Novel Anticancer Fused Pyrazole Derivatives as EGFR and VEGFR-2 Dual TK Inhibitors,Frontiers in Chemistry, vol. 7, pp. 917, 1-12.
Sandeep, S, Priyadarshini, V, Pradhan, D,et al.(2012). Docking and molecular dynamics simulations studies of human protein kinase catalytic subunit alpha with antagonist, The Journal of Clinical and Scientific Research,vol. 1, no. 1, pp. 15-23.
Sasaki, T, Hiroki, K and Yamashita, Y (2013). The role of epidermal growth factor receptor in cancer metastasis and microenvironment, Biomed research International, Vol. 2013, pp. 1-8.
Sung, H, Ferlay, J, Siegel, RL, et al.(2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, CA: A Cancer Journal for Clinicians,vol. 71, no. 3, pp. 209-249.
Toolabi, M, Moghimi, S, Bakhshaiesh, TO, et al.(2019). 6-Cinnamoyl-4-arylaminothienopyrimidines as highly potent cytotoxic agents: Design, synthesis and structure-activity relationship studies,European journal of medicinal chemistry, vol. 185, pp. 111786.
Tsubata Y, Tanino R and Isobe T (2021). Current Therapeutic Strategies and Prospects for EGFR Mutation-Positive Lung Cancer Based on the Mechanisms Underlying Drug Resistance, Cells, vol. 10, no. 11, pp. 3192.
Tu, Y, Yang, YO, Xu, S, et al.(2016). Design, synthesis and docking studies of afatinib analogs bearing cinnamamide moiety as potent EGFR inhibitors,Bioorganic & Medicinal Chemistry,vol. 24, no. 7, pp. 1495-503.
Wang, HC, Yan, XQ, Yan, TL, et al.(2016). Design, Synthesis and Biological Evaluation of Benzohydrazide Derivatives Containing Dihydropyrazoles as Potential EGFR Kinase Inhibitors, Molecules,vol. 21, no. 8, pp. 1012.
Wu, L, Ke, L, Zhang, Z, et al.(2020). Development of EGFR TKIs and Options to Manage Resistance of Third-Generation EGFR TKI Osimertinib: Conventional Ways and Immune Checkpoint Inhibitors, Frontiers in Oncology, vol. 10, pp:2778.
Xiao, Z, Zhou, Z, Chu, C, et al.(2020). Design, synthesis and antitumor activity of novel thiophene-pyrimidine derivatives as EGFR inhibitors overcoming T790M and L858R/T790M mutations, European Journal of Medicinal Chemistry,vol. 203, pp. 112511.

