Department of Microbiology and Biotechnology, C.U. Shah Institute of Life Sciences, Surendranagar, Gujarat, India.
2Department of Post Graduate Diploma in Medical Laboratory, Mehsana Urban
Institute of Sciences, Ganpat University, Mehsana, Gujarat, India.
3Bioinformatics Centre, Birla Institute of Scientific Research, Jaipur, India.
4Department of Science & Technology, Gujarat Council of Science & Technology, Gujarat, India.
Corresponding author email: girishkgoswami@gmail.com
Article Publishing History
Received: 15/09/2021
Accepted After Revision: 21/12/2021
A simple explanation for antimicrobial-resistant opportunistic infections in immunocompromised patients is Klebsiella pneumoniae which gradually being associated in insidious infections globally with high mortality rate. Eight hundred fifty-six antibiotic resistant K. pneumoniae isolates were collected over 3 years period (from different wards and different specimens) from the Microbiology department of C.U. Shah hospital, whose AST checked by Kirby Bauer disk diffusion method. To study AMR genes, virulome, interference of virulence gene with resistance gene, phylogenomic; 6 clinical isolates were proceeded for whole genome sequencing and bio informatics analysis. Klebsiella pneumoniae is a multidrug-resistant (MDR) opportunistic and one of delegate of ESKAPE pathogens groups.
This pathogen causes nosocomial infections, urinary tract infections, liver abscesses, wound infections, meningitis. These strains obtain a multidrug resistant phenotype by way of horizontal transfer of ARG transported by either transposons or plasmids. This transfer is generally facilitated by Integrons. In this study antibiotic resistance profile and antibiotic resistance genes analysis as well as virulence gene of K. pneumoniae strains were investigated. The study was carried out using 853 clinical isolates collected during 3 years from C.U. Shah hospital of Surendranagar. Antibiotic resistance profile test was carried out by the VITEK 2 against 21 antibiotics.
Out of that 6 samples were proceed for DNA extraction, WGS illumina sequencer and analysis of those raw sequences by TORMES pipeline. In this study antibiotic resistance profile included 13 beta lactam antibiotics which classified under 3 class (Penicillin, Cephalosporin, Carbapenem) of beta lactam and in AMR gene study got total 15 different ESBL resistance genes from 6 different klebsiella pneumoniae strain. All these genes detected with more than 90% identity by CARD. (TORMES Pipeline) CTX-M-15, NDM-5, OKP-B-6, PDC-2, OXA-1, OXA-181, OXA-362, OXA-50, OXA-9, SHV-1, SHV-11, SHV-187, TEM-1, TEM-150.
In this study, we’ve analyzed the pattern of antibiotic resistance pattern as a phenotypic characteristic and antibiotic resistance genes as genotypic characteristic and co related the results. As multidrug resistance is a worrying matter, constant observation and regular clinical recognition of resistant bacteria are essential to avoid terrible public health incidents. So, our data should be inferred as a warning for need for prevention and control of the MDR K. pneumoniae in hospital settings.
Antibiotic Resistance, ARG, Klebsiella Pneumoniae, Whole Genome Sequencing
Patel V, Mehta P, Krishnamohan M, Kikani K, Goswami G. Whole Genome Sequencing and Beta-Lactam Resistant Genes in Klebsiella pneumoniae Strains Clinically Isolated at the C.U. Shah Hospital, Gujarat, India. Biosc.Biotech.Res.Comm. 2021;14(4).
Patel V, Mehta P, Krishnamohan M, Kikani K, Goswami G. Whole Genome Sequencing and Beta-Lactam Resistant Genes in Klebsiella pneumoniae Strains Clinically Isolated at the C.U. Shah Hospital, Gujarat, India. Biosc.Biotech.Res.Comm. 2021;14(4). Available from: <a href=”https://bit.ly/31olaRM“>https://bit.ly/31olaRM</a>
Copyright © Patel et al., This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) https://creativecommns.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
In the treatise Antibiotic resistance, a universal fact is broadly defined which linked with therapeutic failure and increase growth rate in sickness as well as death (Julian et al. 2017). Spreading of resistance in Enterobacteriaceae family is because of swapping the antimicrobial resistance gene amongst the species in hospital environment by plasmid, transposons and integrons like MEGs (Valentina et al. 2019; Kelly et al. 2020).
This can be observed in gram +Ve and -Ve bacteria which is classified by WHO as critical priority pathogens. K. pneumoniae is one of the gram -ve pathogenic bacteria which is in red zone of WHO. As per literature total 8 integrons class are available in which first 3 are specifically connected with antibiotic resistance (Sara et al. 2018; Fernanda et al. 2018; Mehdi et al. 2021).
Klebsiella pneumoniae is gradually intrusive virulent pathogen with antibiotic resistance genes like extended-spectrum β-lactamases (ESBLs), AmpCs and carbapenemases in Enterobacteriaceae family. So, to elaborate the resistome and its mechanism as well as MGEs linked with them and phylogenomic of K. pneumoniae, we carried out this study (Hongmao et al. 2021).
MATERIAL AND METHODS
In the present study, total 856 numbers of clinical specimens in the form of different samples (urine, blood, sputum, CSF, swab, pus etc.) from different wards (medicine, surgery, orthopedics, pulmonary medicine, ENT, pediatrics) of C.U Shah medical college were collected, processed and examined at microbiology department of C.U. Shah hospital, Surendranagar from duration of January (2016) to December (2018) (Ethical approval taken-CUSMC/IEC(HR)/APPROVAL-13/2018/1288).
By morphological characteristics on MacConkey agar (HI Media) clinical isolates of Klebsiella (large mucoid colonies) were identified and isolated. All isolates were further proceeded for gram staining for primary confirmation of the strain. (Clinical and Laboratory Standard Institute, 27th edition). Antibiotic susceptibility testing was performed for all bacterial isolates by modified Kirby Bauer disk diffusion method as per Clinical and Laboratory Standards Institute (CLSI) guidelines.
A suspension equivalent to 0.5 McFarland standards was prepared from single isolated colony. A swab was dipped in suspension and streaked over surface of a Mueller-Hinton agar plate. (Clinical and Laboratory Standard Institute, 27th edition). Obtained Isolates were subjected to susceptibility testing for the following classes of antibiotics: Amikacin, Amoxicillin, Amoxicillin/Clavulanic Acid, Cefepime, cefixime, cefoperazone, cefoperazone/Sublactum, cefotaxime, ceftazidime, ceftriaxone, chloramphenicol, ciprofloxacin, co-trimoxazole, colistin, gentamycin, imipenem, levofloxacin, ofloxacin, piperacillin/tazobactam, tetracycline, tobramycin. (Clinical and Laboratory Standard Institute, 27th edition).
The DNA extraction was performed using the HiPurATM Bacterial Genomic DNA Purification Kit MB505. Samples were processed according to kit the instructions for optimum yield. From each isolated DNA sample, 2 µL was used to determine the 260:280 ratios for DNA quality determination and to estimate the DNA concentration using the Nano Drop 1000 spectrophotometer and Qubit Fluorometer. At least 500 ng of DNA for each sample was sent for further sequencing analysis.
Samples for WGS were processed at the Med Genome Labs Pvt. Ltd., Bangalore 560 099, India. Different qualitative and quantitative control steps were taken for samples at various stages of the library construction (i.e., initial receipt, post-shearing, post-library construction) as per GLP criteria. The sequencing center provided libraries in Fastq format.
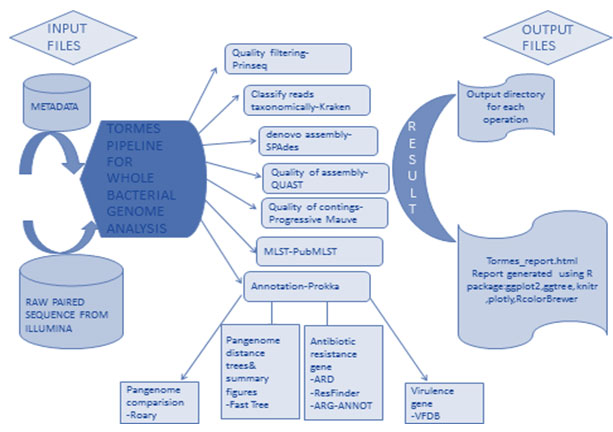
The raw sequences were assembled and annotated with TORMES. It was an open-source, user-friendly pipeline for whole bacterial genome sequencing (WGS) analysis directly from raw Illumina paired-end sequencing data. TORMES functioned with all bacterial WGS dataset, despite the number, source, or species. TORMES pipeline is freely available in https://github.com/nmquijada/tormes, with a manual how to use it. TORMES pipeline and all the required software and dependencies can be automatically installed by using Conda (Quijada et al. 2019).
RESULTS AND DISCUSSION
The study about this suggest that genes linked with antimicrobial resistance (AMR) are each strong and from different sub populations (Holt et al. 2015; Wyres et al. 2019). So, there is a possibility of a dangerous strains which are highly pathogenic and resistant to all or almost available antibiotics. (Gu et al. 2017; Lam et al. 2019; Mehdi et al. 2021).
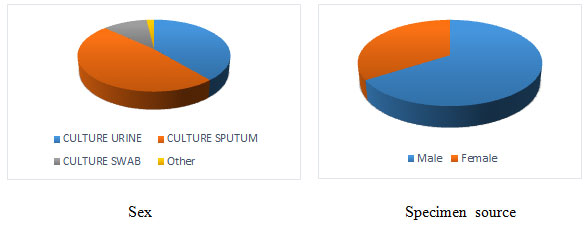
Figure 1: From the 3 years data of patients demonstrate the frequency of different
type specimen sources and ratio of infected male and Female
All the isolates obtained from the samples were detected phenotypically and genotypically as K. pneumoniae. All 6 isolates were phenotypically positive for ESBL production. As per the study we found isolate 027 was MDR, 678 was ESBL (+HL AmpC) and carbapanamase positive, 999 was ESBL (+HL AmpC) and carbapanamase positive, 968 was ESBL (+HL AmpC) and carbapenemase positive, 982 and 996 were ESBL positive. Penicillin class antibiotics carried same MIC in all the isolates but variable MIC demonstrated in carbapenem class antibiotics specifically with Imipenem. Four isolates were non susceptible for cefuroxime, cefotaxime, ceftriaxone and cefepime facing same MIC value (Mehdi et al. 2021).
Figure 2: Flow charts show the summary of TORMES pipeline
Phenotypic resistance data matched mostly with the genomic results with some exceptions in this study. In this Phenotypic study included 13 beta lactam antibiotics which classified under 3 class (Penicillin, Cephalosporin, Carbapenem) of beta lactam and in genotypic result got total 15 different ESBL resistance genes from 6 different Klebsiella pneumoniae strain. All these genes detected with more than 90% identity by CARD. (TORMES Pipeline) CTX-M-15, NDM-5, OKP-B-6, PDC-2, OXA-1, OXA-181, OXA-362, OXA-50, OXA-9, SHV-1, SHV-11, SHV-187, TEM-1, TEM-150. (Figure-2) (Table 1) (Mehdi et al. 2021).
Table 1. beta lactam resistance genes present in samples
| GENE | Sample1(027) | Sample2(678) | Sample3(968) | Sample4(982) | Sample5(996) | Sample5(999) |
| CTX-M-15 | 100 | 100 | ||||
| NDM-5 | 100 | |||||
| OKP-B-6 | 99.88 | |||||
| OXA-1 | 100 | |||||
| OXA-181 | 100 | 100 | ||||
| OXA-50 | 100 | |||||
| OXA-9 | 100 | |||||
| PDC-2 | 100 | |||||
| SHV-1 | 100 | 100 | ||||
| SHV-11 | 100 | |||||
| SHV-187 | 100 | |||||
| TEM-1 | 100 | |||||
| TEM-150 | 100 |
A total of 18 virulence genes were identified in all 6 strains. OmpA, fimE, fimA, ents, fepD, fepC, fepA, entA, entB, entE, fepB, yagV/ecpE, yagW/ecpD, yagX/ecpC, yagY/ecpB, yagZ/ecpA, yagK/ecpR. The search of virulence genes was performed by screening of the draft genome against Virulence Factors Data Base by using Abricate (Chen et al. 2005; Morales et al. 2021).
It is to be noted that these are the genes which are present in all the isolates either resistant or susceptible for different antibiotic. As per VFDB fimE and fimA are type one fimbriae protein and expressed in 90% of clinical K. pneumoniae isolates as well as entA, entB and entE are responsible for ion acquision. Whereas fepA, fepB, fepC, and fepD are siderophore. (Morales et al. 2021).
Phylogenomic and MLST Analysis:
Figure 3: Phylogenetic diversity
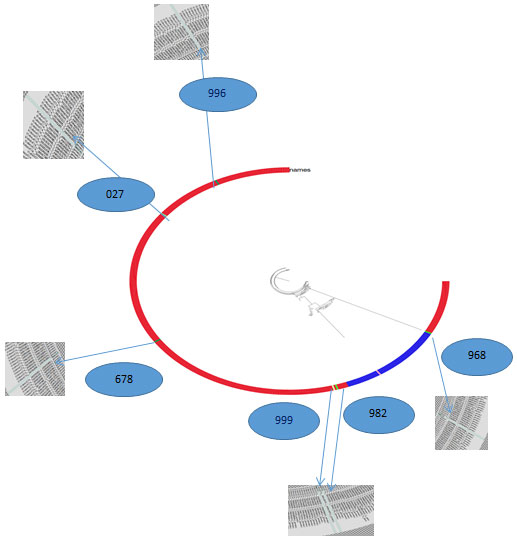
Along with analysis of whole genome sequence, for evolutionary study phylogenomic analysis was carried out for selected K. pneumoniae isolates by Fast Tree. Which infers approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences (http://www.microbesonline.org/fasttree/). In this study it is done with 365 Klebsiella species. (NCBI dataset).
K. pneumoniae isolates exhibited important phylogenetic diversity (Figure-3), with the whole-genome phylogenetics presenting higher resolution than the MLST typing scheme. For example, K5 (982) was phylogenetically closer to K7 (999) than other K3(968) strains, which were themselves found on different branches. Remarkably, K6 (027), K1 (996) had phylogenetically distant from other strain but they both somewhat closer within the collection.
Multilocus Sequence typing (MLST) was performed according to the protocol described by PubMLST database (for Klebsiella pneumoniae), Github https://github.com/tseemann/mlst in Conda environment on the whole genome assemblies. The MLST profile derived from the following housekeeping genes: gapA (glyceraldehyde 3-phosphate dehydrogenase), infB (translation initiation factor 2), mdh (malate dehydrogenase), pgi (phosphoglucose isomerase), phoE (phosphorine E), rpoB (beta subunit of RNA polymerase) and tonB (periplasmic energy transducer).
(Github https://github.com/tseemann/mlst). Molecular mechanisms of resistance and virulence spreading in clinical K. pneumoniae strains circulating in hospitals were illustrated and noted to be ornately capable with various factors of resistance, virulence and mobile-genetic elements. Especially, the isolates were MDR to several clinically important antibiotics except for reserved ones such as the colistin. Presence of the MDR strain and the strains which are sensitive to only one or two antibiotics in specimens from the various patients in hospital make this a very perturbing result (Nontombi et al. 2020).
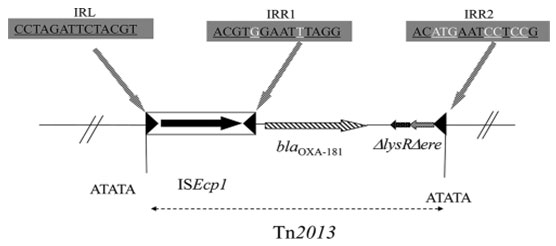
The presence of the CTX-M, TEM, OXA, and SHV ESBL genes in analogous genetic frameworks have been illustrated previously in same and different species or different bacteria from same family in India and global isolates. The presence of these ARGs in the similar genetic environment and on the similar plasmid replicons throughout similar and dissimilar species everywhere in the world effectively indicates the clonal and plasmid-mediated distribution of these ARGs (Reza et al. 2019; Nontombi et al. 2020). Precisely, ISEcp1 plasmid have been revealed to mobilize and facilitate the global spread of CTX-M-15. Including the various CTX-M variants, CTX-M-15 was the major and it was first stated from the Indian subcontinent (Roghieh et al. 2021).
Comparison of gene sequences in the GenBank database revealed homology of the positions 1436 to 2312 in E. coli with CTX-M-15 (100% identity, position 2719 to 3594; accession number AY044436:1436-2312). In our study isolate 027 and 678 which carried this gene were resistant to cefotaxime, ceftriaxone, cefaperazone/sublactum (all from 3rd generation) and cefepime from 4th generation with >=64 MIC. It was located with the ISEcp1 sequence at upstream of 5’ end on the plasmid. (Ramesh et al. 2017; Roghieh et al. 2021).
Figure 4: (Karim A., et al 2011) (https://www.ncbi.nlm.nih.gov/nuccore/15636728/?from=1436&to=2312&report=graph)

We reported a similar prevalence of OXA and SHV β–lactamase genes in these isolates, make sure that this apparently chromosomally encoded gene, is almost present in K. pneumoniae species. The strong catalog of ARGs in these isolates indicate the existence of one or more plasmids. OXA-1 plasmid-mediated β-lactamases among K. pneumoniae strains indicated the resistance against beta lactam antibiotics especially ampicillin and piperacillin.
Several plasmid-encoded beta lactamases are on multi resistance transposable elements. The OXA-1 beta-lactamase gene is part of Tn2603. Comparison of gene sequences in the GenBank database revealed homology of the positions 1400 to 2231 in Klebsiella pneumoniae with OXA-1(100 % identity, position 274 to 1104; accession no. JN420336.1:1400-2231) (Madhan et al. 2014; Roumayne et al. 2019; Nontombi et al. 2020).
Figure 5: (Villa, L., et al 2012)(https://www.ncbi.nlm.nih.gov/nuccore/JN420336.1?report=graph&from=1400&to=2231)

Gene OXA-9 was originally found within the structure of Tn1331, a multi resistance transposon first isolated from a clinical Klebsiella pneumoniae strain. Comparison of gene sequences in the GenBank database revealed homology of the positions 1 to 826 in Klebsiella pneumoniae with OXA-9(100 % identity, position 130 to 954; accession no. M55547:1-826). Both OXA-1 and OXA-9 are the part of transposon which is part of plasmid but easily move from plasmid to plasmid and plasmid to chromosome and vice versa which were carrying antibiotic resistance genes (Bojorquez et al. 1998; Navdezda et al. 2019; Katerina et al. 2021).
Figure 6: Tolmasky M.E, et al 1993) (https://www.ncbi.nlm.nih.gov/nuccore/M55547.1?report=genbank)
Figure 7: (Potron et al. 2011) (https://www.ncbi.nlm.nih.gov/nuccore/JN205800.1?report=graph)
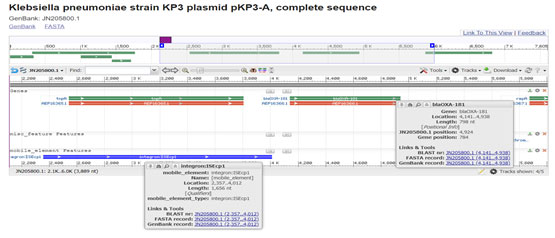
Figure 8: (Potron et al. 2011)
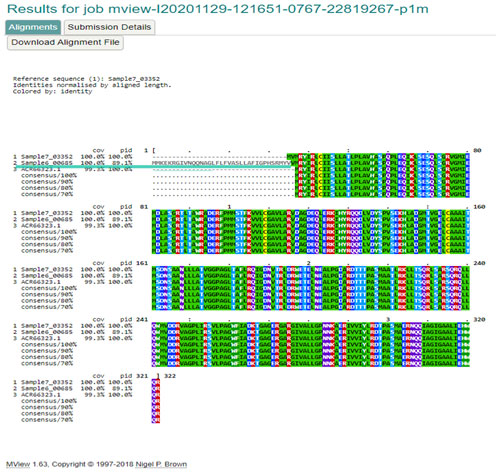
Comparison of gene sequences in the GenBank database revealed homology of the positions 77 to 938 (isolate-996, 999), in Klebsiella pneumoniae with SHV-1 (100 % identity, position 296087 to 296947(996), 254 to1114 (999); accession no FJ668814:77-938). SHV-1 was present in isolate 996 and 999. Phenotypically isolate 999 show resistance towards all the 3 class of beta lactam antibiotics. But isolate 996 phenotypically susceptible for all beta lactam antibiotics though there was presence of SHV-1. Upstream 5’ UTR sequence detected in isolate 996 which was under lined in figure -9 (Isabel et al. 2021).Hypothetically we can predict this UTR sequence not allow the gene expression.
Figure 9: Alignment of gene sequence by clustal omega
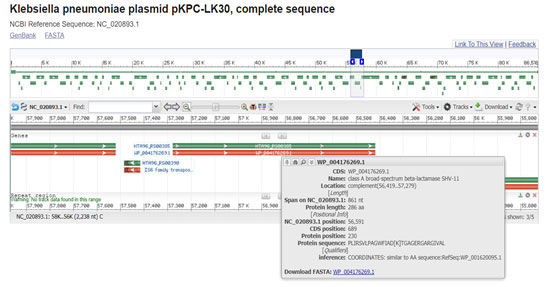
SHV-11 is a broad-spectrum beta-lactamase found in E. coli, Klebsiella pneumoniae, Proteus mirabilis, and Shigella dysenteriae. It is differing from SHV-1 only at amino acid position 35, instead of L(Leu) SHV-11 carried Q (Gln). Comparison of gene sequences in the GenBank database revealed homology of the positions 74 to 935 with SHV-11 (99.77 % identity, position 157681 to 158541; accession no. X98101:74-935). It was present in isolate 678 and resistant to all beta lactam antibiotics which used in our study (Anais et al. 2011; Isabel et al. 2021).
Figure 10: (Chen, Y.T et al. 2014) (https://www.ncbi.nlm.nih.gov/nuccore/NC_020893.1?report=graph&from=55729&to=57966&strand=true)
TEM-1 is the commonly encountered and one of the most well-known beta-lactamase in gram-negative bacteria confirmed resistance against penicillins and generally first-generation cephalosporin. All TEM variants differ in amino acid sequence (in general by one to five substitutions) and many of them differ in resistance phenotype (the degree of resistance they confer to different antibiotics). In this study TEM-1 was detected in isolate 678 and TEM-150 was in isolate 027 (Salverda et al. 2010; Nadezhda et al. 2021).
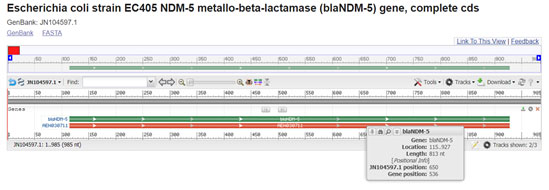
NDM-5 new delhi metallo beta lactamase was detected in isolate 678 only. It resembles resistance against penicillin, cephalosporin and carbapenem class antibiotics. In the study of Hazim et al. in 2016 and Lauren et al. in 2019 NDM-5 was responsible for the resistance against beta lactam antibiotics. If we compare gene sequence in the GenBank database, it revealed homology of the positions 115-928 in E. coli with NDM-5 (100% identity, position 457 to 1569; accession number JN104597:115-928) (Wang et al. 2020).
Figure 11: (Hornsey et al. 2011)(https://www.ncbi.nlm.nih.gov/nuccore/JN104597.1?report=graph)
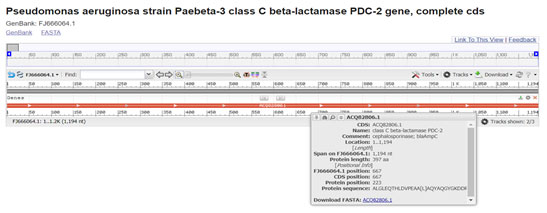
PDC-2 is a class-C ESBL generally present in Pseudomonas aeruginosa express resistance against cephalosporin and carbapenem class antibiotics. It is most frequently AmpC-type variant. We detected it in isolate 968. Comparison of gene sequences in the GenBank database revealed homology of the positions 1 to 1195 in Pseudomonas aeruginosa with CTX-M-15 (99.58 % identity, position 734 to 1927; accession number FJ666064:1-1195) (Zain et al. 2020).
Figure 12: (Rodriguez-Martinez et al. 2009) (https://www.ncbi.nlm.nih.gov/nuccore/FJ666064.1?report=graph)
CONCLUSION
The findings of the present study determine that with the increasing worldwide occurrence of antibiotic resistant pathogens, knowing how bacteria adapt and evolve to drug stress is enormously important. This study showed the distribution of the possible resistome for Klebsiella pneumoniae isolates through WGS analysis. The presence of specific AMR genes represents a development towards higher MICs values for β-lactams anti microbials.
The results of this work showed the association between phenotypic resistance and the number of exclusive AMR gene sequences. There were difference and similarity in the phenotypic and genotypic resistance between bacterial species. Some specific AMR genes found in these genomes could be potentially invented from horizontal transfer of mobile elements. In general, more studies are needed to search the probable sources of these AMR genes.
Conflict of Interests: Authors declare no conflict of interests to disclose.
REFERENCES
Almeida F, Seribelli AA (2018). Phylogenetic and antimicrobial resistance gene analysis of Salmonella typhimurium strains isolated in Brazil by whole genome sequencing. PLoS ONE 13(8): e0201882.
Bojorquez D, Belei M, Delira SF et al. (1998). Characterization of OXA-9, a beta-lactamase encoded by the multi resistance transposon Tn1331. Cell Mol Biol (Noisy-le-grand) 44(3): 483-491.
Brinkac LM, White R, D’Souza R et al. (2019). Emergence of New Delhi Metallo–Lactamase (NDM-5) in Klebsiella quasipneumoniae from Neonates in a Nigerian Hospital. mSphere 4: e00685-18.
Carvalho I, Chenouf NS, Carvalho JA et al. (2021). Multidrug-resistant Klebsiella pneumoniae harboring extended spectrum β-lactamase encoding genes isolated from human septicemias. PLoS ONE. 16:(5), e0250525.
Chen YT, Lin JC, Fung CP et al. (2014). KPC-2-encoding plasmids from Escherichia coli and Klebsiella pneumoniae in Taiwan J Antimicrobe Chemother 69 (3), 628-631.
Chudejova K, Kraftova L, Marchett VM et al. (2021). Genetic Plurality of OXA/NDM-Encoding Features Characterized from Enterobacterales Recovered from Czech Hospitals. Front. Microbiol. 641415.
Clinical and Laboratory Standard Institute (2017). Performance standards for antimicrobial susceptibility testing, (27).
Ferreira RL, Silva BD, Rezende GS et al. (2019). High Prevalence of Multidrug-Resistant Klebsiella pneumoniae Harboring Several Virulence and β-Lactamase Encoding Genes in a Brazilian Intensive Care Unit fmicb.2018.03198.
Galata V (2019). Integrating Culture-based Antibiotic Resistance Profiles with Whole-genome Sequencing Data for 11,087 Clinical Isolates. Genomics Proteomics Bioinformatics 17: 169–182.
Golsha R, Montazeri M, Razagh N et al. (2021). Frequency of Beta-Lactamase Antibiotic Resistance Genes in Escherichia Coli and Klebsiella pneumoniae. Ethiopian Journal of Health Sciences. 31: 5.
Gu D, Dong N, Zheng Z et al. (2017). A fatal outbreak of ST11 carbapenem resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect. Dis. 3099, 1–10.
Hazim O, Khalifa SS, Elnahriry AM, et al. (2016). Emergence of Plasmid-Mediated Colistin Resistance Gene mcr-1 in a Clinical Escherichia coli Isolate from Egypt. Antimicrob Agents Chemother. 60(5):3249-50.
Holt KE and Zadoks RN (2015). Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl Acad. Sci. USA 112, E3574–E3581.
Hornsey M, Phee L and Wareham DW (2011). A Novel Variant, NDM-5, of the New Delhi Metallo-beta-Lactamase in a Multidrug-Resistant Escherichia coli ST648 Isolate Recovered from a Patient in the United Kingdom Antimicrob. Agents Chemother. 55 (12), 5952-5954.
Karim A, Poirel L, Nagarajan S et al. (2001). Plasmid-mediated extended-spectrum beta-lactamase (CTX-M-3 like) from India and gene association with insertion sequence ISEcp1 FEMS Microbial. Lett. 201 (2), 237-241.
Kashefieh M, Hosainzadegan H, Baghbanijavid S et al. (2021). The Molecular Epidemiology of Resistance to Antibiotics among Klebsiella pneumoniae isolates in Azerbaijan, Iran. Journal of Tropical Medicine. ID 9195184:2021.
Kelly LW, Lam MMC, Holt KE et al. (2020). Population genomics of Klebsiella pneumoniae. Nature Reviews Microbiology 1579-019-0315-1.
Lam MMC and Kelly LW (2019). Convergence of virulence and MDR in a single plasmid vector in MDR Klebsiella pneumoniae ST15. J. Antimicrob. Chemother. 74, 1218–1222.
León AM and Opazo-Capurro CC et al. (2021). Hypervirulent and hypermucoviscous extended-spectrum β-lactamase-producing Klebsiella pneumoniae and Klebsiella variicola in Chile. VIRULENCE.12(1), 35-44.
Liu H, Lin H, Sun Z et al. (2021). Distribution of β-Lactamase Genes and Genetic Context of blaKPC-2 in Clinical Carbapenemase-Producing Klebsiella pneumoniae Isolates. Infect Drug Resist. 14: 237–247.
Lomonaco S, Crawford MA, Lascols C et al. (2018). Resistome of carbapenem- and colistin resistant Klebsiella pneumoniae clinical isolates. PLoS ONE 13(6): e0198526.
Maurya N, Jangra M, Tambat R et al. (2019). Alliance of Efflux Pumps with b-Lactamases in Multidrug-Resistant Klebsiella pneumoniae Isolates. Microbial drug resistance, Volume 25.
Mbelle NM, Feldman C, Sekyere JO et al. (2020). Pathogenomics and Evolutionary Epidemiology of Multi-Drug Resistant Clinical Klebsiella pneumoniae Isolated from Pretoria, South Africa. Scientific Reports, 10:1232.
Nachimuthu R (2017). The distribution of carbapenem- and colistin-resistance in Gram-negative bacteria from the Tamil Nadu region in India. Journal of Medical Microbiology. 66:7.
Nadezhda K, Fursova EI, Astashkin ON, et al. (2021). Multidrug-Resistant Klebsiella pneumoniae Causing Severe Infections in the Neuro-ICU. Antibiotics. 10:8.
Potron A, Nordmann P, Lafeuille E et al. (2011). Characterization of OXA-181, a Carbapenem-Hydrolyzing Class D β-Lactamase from Klebsiella pneumoniae. Antimicrobial agents and chemotherapy, 4896–4899.
Potron A, Nordmann P, Lafeuille E et al. (2011). Characterization of OXA-181, a Carbapenem-Hydrolyzing Class D {beta}-Lactamase from Klebsiella pneumoniae. Antimicrob. Agents Chemother. 55 (10), 4896-4899.
Quijada NM, Rodríguez-Lázaro D, Eiros JM et al. (2019). TORMES: an automated pipeline for whole bacterial genome analysis. Bioinformatics, 35(21), pp.4207-4212.
Ranjbar R, Kelishadrokhi AF and Chehelgerdi M (2019). Molecular characterization, serotypes, and phenotypic and genotypic evaluation of antibiotic resistance of the Klebsiella pneumoniae strains isolated from different types of hospital-acquired infections. Infect Drug Resist. 12: 603–611.
Rodriguez-Martinez JM, Poirel L and Nordmann P (2009). Extended-spectrum cephalosporinases in Pseudomonas aeruginosa Antimicrobe. Agents Chemother. 53 (5), 1766-1771.
Salverda ML, Visser AGMJD, Barlow M et al. (2010). Natural evolution of TEM-1 β-lactamase: experimental construction and clinical relevance. FEMS Microbiol. Rev. 34(6):1015-36.
Sugumar M, Kumar KM, Manoharan A et al. (2014). Detection of OXA-1 β-Lactamase Gene of Klebsiella pneumoniae from Blood Stream Infections (BSI) by Conventional PCR and In-Silico Analysis to Understand the Mechanism of OXA Mediated Resistance. PLoS ONE 9(3): e91800.
Tolmasky ME and Crosa JH (1993) Genetic organization of antibiotic resistance genes (aac(6′)-Ib, aadA, and oxa9) in the multi resistance transposon Tn1331 Plasmid 29 (1), 31-40.
Vélez JR, Carlos MCJ et al. (2017). Whole-genome sequence analysis of antimicrobial resistance genes in Streptococcus uberis and Streptococcus dysgalactiae isolates from canadian Dairy herds Front. Vet. Sci. 4:63.
Villa L, Poirel L, Nordmann P et al. (2012). Complete sequencing of an IncH plasmid carrying the blaNDM-1, blaCTX-M-15 and qnrB1 genes J. Antimicrob. Chemother. 67 (7), 1645-1650.
Wang H, Li X and Liu B (2020). Occurrence and characterization of KPC-2-producing ST11 Klebsiella pneumoniae isolate and NDM-5-producing Escherichia coli isolate from the same horse of equestrian clubs in China. Transboundary and Emerging Diseases.68:(2), 224-232.
Wyres KL, Wick RR, Judd LM et al. (2019). Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 15, e1008114.
Zain I, Alamarat J, Babic TT et al. (2020). Long-Term Compassionate Use of Cefiderocol to Treat Chronic Osteomyelitis Caused by Extensively Drug-Resistant Pseudomonas aeruginosa and Extended-Spectrum-β-Lactamase-Producing Klebsiella pneumoniae in a Pediatric Patient. Antimicrobial Agents and Chemotherapy.64:(4).

