1Department of Biotechnology, University of Calicut, Calicut University PO, Malappuram 673 635, India
2Bioinformatics Infrastructure Facility, Department of Biotechnology, University of Calicut, Calicut University PO, Malappuram 673 635, India
Article Publishing History
Received: 06/09/2016
Accepted After Revision: 29/09/2016
MicroRNAs are endogenous, non-translated, small RNAs of ~21 nucleotides that are processed from stem-loop regions of long transcripts. MicroRNA is responsible for regulation of gene expression of diverse aspects of plant development at the post-transcriptional level. Coconut is one of the major perennial crop, that has wide commercial importance. The identification of novel miRNAs from non-sequenced genome of plants can be done by comparative genomics approach using expressed sequence tags (ESTs) following filtering criteria based on structural features.In an attempt to identify potential miRNAs from Cocos nucifera,1008 ESTs were employed for homology based search to reported viridiplantae miRNAs. The candidate miRNAs were used for secondary structure prediction that led to the identification of one novel miRNA from mir2673 family. The gene targets predicted miRNA shows crucial role in regulation of auxin signaling pathway, transcription factors, abiotic stress, retrotransposons etc. The outcomes of this study will considerably enhance the scope to understand the role of miRNAs in C.nucifera.
Cocos Nucifera L. ,Expressed Sequence Tags, Microrna, Microrna Targets, Rna Secondary Structure.
Vivek A. T, Moossa F. Identification of Novel Micro RNAs and their Targets in Cocos Nucifera–A Bioinformatics Approach. Biosc.Biotech.Res.Comm. 2016;9(3).
Vivek A. T, Moossa F. Identification of Novel Micro RNAs and their Targets in Cocos Nucifera–A Bioinformatics Approach. Biosc.Biotech.Res.Comm. 2016;9(3). Available from: https://bit.ly/2N93IWR
Introduction
MicroRNAs are a set of endogenous non-coding RNAs of ~21 nucleotides in length, which has vital role in the transcriptional and post-transcriptional regulation of gene expression (Zhang et al., 2006). These act as an important regulator in various processes of development, cell signaling and stress conditions (Millar et al., 2005;Sunkar et al., 2006; Khraiwesh et al., 2012). The expression of single gene may be controlled by multiple miRNAs and single miRNA can have multiple gene targets (Dehury et al., 2013).
The plant miRNA genes are present in both introns and exons which are transcribed to pre-miRNA(primary miRNA) transcripts and there exists capped structures as well polyA tail that are processed to form stem loop structures. The mature miRNAs are produced from these pre-miRNA transcripts by DCL1 (Dicer like-1) and endonuclease III-like enzyme,is exported via HASTY 5 to the cytosol. The target gene expression is regulated by RNA-induced silencing complex (RISC) containing mature miRNA incorporated into it (Bartel., 2004). In plants, the first miRNA was reported in Arabidopsis thaliana ,following which several thousands of novel miRNAs has been discovered in plants by computational and experimental techniques(Reinhart et al., 2002; Griffiths-Jones and Sam., 2006; Zhang et al., 2016; Akter et al., 2014).
The miRNAs in plants represent a high degree of perfect or nearly perfect complementary to their targets(Rhoades et al., 2002). This highly conservative character of plant miRNAs promises in screening of homolog sequences of miRNAs from nucleotide archives of different plant kingdoms using in silico techniques based on homology (Das et al., 2010). Many conserved miRNAs have been screened in various model and non-model plants by using the genomic data and homology based computational techniques, as exemplified in the study which resulted in identification of 682 miRNAs in 155 diverse plant species (Sunkar et al., 2008). Computational predictions of miRNAs has also been reported in many of the non-model crops such as garlic, ginger, cannabis and basil (Panda et al., 2014; Singh et al., 2014; Das et al., 2015 and Singh et al., 2016).
Cocos nucifera.L. (2n=2x=32), a member of Arecaceae family , is one of the commercially important palms. Being a perennial tree, it is cultivated widely across the world for its versatile utilities uses from food to cosmetics (Govaerts., 2003). Since the available genetic resources are less, comprehensive analysis on the available data in online repositories has to be exercised. At present, no miRNA has been predicted using the available ESTs for C.nucifera. Hence, in this study, an in silico approach for screening of potential miRNAs was performed using homolog search of Viridiplantae miRNAs (miRBase) against the expressed sequence tags. The elimination of coding sequences and use of non-coding ESTs for secondary structure of the precursor miRNA was predicted using MFOLD which led to the identification of novel miRNA. Finally, the predicted novel miRNA was used to find the target genes from Arabidopsis thaliana transcripts.
Material And Methods
The reported 8,496 miRNAs belonging to Viridiplantae were downloaded from miRBase (Released 21: June, 2014) and clustered by CD- HIT-EST, with threshold value of 100 (Li et al., 2006). From the clustered sequences of 3777, non-redundant miRNAs were selected which in turn used as reference miRNAs for finding the homologs in C. nucifera to create a local nucleotide sequence database. Publically available, 1008 ESTs were downloaded from EST database, NCBI (www.ncbi.nlm.nih.gov/dbEST/) to represent query sequences for miRNA
prediction.
The alignment tool NCBI Blast+ was used for conserved miRNA prediction of Cocos nucifera ESTs by making local database of miRNA after retrieving the representative sequences of miRNA (Cock et al., 2015). The secondary structures of pre-miRNAs were performed through online version of MFOLD (Zuker and Michael., 2003). The putative target genes for the predicted miRNA of coconut were identified using plant small RNA psRNATarget Web Server (Dai et al., 2011). The circos plot was drawn using online Circoletto tool(Darzentas et al.,
2010).
The EST sequences were taken to perform blastn search against locally setup Viridiplantae miRNA database downloaded from miRBase. The blast search was performed with following parameters: (a) e-value threshold<0.01 (b) mismatches=0-2. The obtained hits with less than 3 nucleotide mismatches and without gap were selected to extract the precursor sequences (pre-miRNA). The method of a sliding window of 100 nt in size from ~80nt upstream and ~80nt nucleotide downstream of the mature miRNA were set to pick the best miRNA precursors (Singh and Nagaraju., 2008).
The secondary structures were predicted using web based MFOLD tool with following criteria:
(a) linear RNA sequence
(b) folding temperature fixed at 37 o C
(c) ionic conditions of 1 M NaCl without divalent ions
(d) percent sub-optimality number of 5
(e) maximum interior/bulge loop size of 30
(f) energy dot plot was turned on and the other parameters were set as default.
After prediction of the secondary structure of the precursor sequence of potential miRNA homologs, the following criteria were setup to select potential miRNA as described by Zhang in 2005 (Zhang et al., 2005): (a) A minimal length of the pre-miRNA: 60 nt. (b) The pre-miRNA must be into appropriate stem loop hairpin secondary structure. (c) The position of mature miRNA sequence should be in one arm of the hairpin structure.(d) The mature miRNA sequence and its opposite miRNA strand should not have more than 6 nt mismatches. (e)The total A+U % should be in the range of 30-70%. (f) should have higher minimal folding free energy index (MFEI) and negative minimal folding free energy (MFE) to distinguish the miRNA secondary from other small RNAs. (f) The MFEI and adjusted MFE(AMFE) was calculated using the following equations:
MFEI =[(AMFE/(G+C)% ]
AMFE=[MFE÷ length of precursor miRNA] X100
The perfect complementarity or near complementarity permits the identification of miRNA targets. The C. nucifera miRNA targets were identified through homolog search by subjecting mature miRNA sequence as query against Arabidopsis thaliana transcript library with removed miRNA gene set(ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/TAIR10_blastsets/TAIR10_cdna_20101214_updated) using psRNAtarget webserver. The following parameters were employed in prediction of miRNA targets :
(a) No.of target genes for each small RNA:10
(b) Maximum mismatch at complementary site:≤2 without any gaps
(c) Maximum exception of 2.0
(d) Target accessibility-allowed maximum energy to un-pair the target site (UPE): 25
(e) Range of central mismatch leading to translation inhibition: 9–11 nt
(f) Flanking length around the target accessibility analysis: 17 bp upstream and 13 bp in downstream length of complementarity score: 20
The entire workflow for the prediction of coconut miRNAs is illustrated in Fig.1
Results And Discussion
The conserved microRNAs in plants among different species including monocot and dicot are involved in various biological activities (Yang et al.,2007). The novel miRNA in C.nucifera was predicted using homology based in silico approach. The known miRNA from mirBase and the available C.nucifera ESTs were performed BLAST to obtain a total of 13 homologs after elimination of repeated sequences. Thereafter, 13 ESTs were screened for coding and non-coding sequences, which resulted in 9 non-coding miRNAs, out of which strong non-coding sequences were taken based on the basis of coding potential score (Ref. Table 1).
 |
Figure 1: Workflow of Coconut miRNA prediction |
| Table 1: List of coding and non-coding ESTs | ||
| ID | Coding/Non-coding | Coding potential score |
| 573332413 | coding | 2.81173 |
| 573332205 | noncoding | -1.34675 |
| 573332140 | coding | 0.197992 |
| 573332096 | noncoding | -1.39801 |
| 573332024 | noncoding | -0.82414 |
| 573332021 | noncoding | -1.21493 |
| 573331999 | noncoding | -1.05615 |
| 573329619 | noncoding | -1.26066 |
| 573329529 | noncoding | -1.23343 |
| 573329506 | coding | 0.746432 |
| 573329506 | coding | 0.746432 |
| 323150074 | noncoding | -1.08997 |
| 323150034 | noncoding | -1.39945 |
After circumspectly considering nucleotide BLAST and CPC tool results, we were able to identify the nine unique EST sequences that showed homology with the known the miRNAs in mirBase.These unique ESTs were subjected for secondary structure prediction and the results attained were investigated for appropriate precursor miRNA and its respective stem-loop structure. On applying the filtering criteria of secondary structure prediction, one novel miRNA was found (Fig. 2 and Fig. 3). The miRNA represented mir 2673 family with a precursor length of 99nt, which justifies the previous reports that miRNA precursors length varies from 60-400 nucleotides (Zhang et al., 2006). If the calculated MFEI is greater than 0.67, then there is a possibility for precursor miRNA (Yin et al., 2008). MFEI of the predicted coconut miRNA was found to be 0.80, which supports this criterion. The AMFE calculated for the coconut miRNA was 50.2 -kcalmol-1 which follows the reported average AMFE (45.93±9.43 -kcalmol-1) of 513 precursor miRNA of plants (Zhang et al., 2006) (Ref. Table 2).
 |
Figure 2 |
 |
Figure 3: EST sequence containing pre-miRNA(yellow color higlighted) and mature miRNA(blue color highlighted) |
| Table 2: Details of the predicted miRNA | |
| Length of EST sequence | 1008 |
| Length of precursor miRNA | 99nt |
| Length of mature miRNA | 19nt |
| Precursor miRNA coordinates | 74-172 |
| Mature miRNA coordinates | 135-153 |
| Family of miRNA | mir 2673 |
| MFE | 49.70 -Kcal/mol |
| MFEI | 0.80 |
| AMFE | 50.2020 -Kcal/mol |
| Nucleotide mismatch | 1 |
| Number of each nucleotides in pre-miRNA sequences | A-14
T/U-23 G-36 C-26 |
| (A+T/U)% | 37.38% |
| (G+C)% | 62.62 % |
| Precursor miRNA sequence | GGGUUUUUGGGUCUCGCUCCCUUCUU
CUGCCCUUCGCCUCUCGGCGUAGGUCA GGUGAGGCGAAGACGAAGAGGAAGAG GAGGAGCCCUCGCCGUGUGG |
| Mature miRNA sequence | GAAGACGAAGAGGAAGAGG |
| Star miRNA sequence | CUUCCCGUCCUUCUUCCC |
So far, the predicted miRNA family is reported only in Medicago truncatula and therefore progress has to be made in understanding the role of this novel miRNA (Lelandais-Brière et al., 2009). As proposed by miRBase, the new miRNA of coconut is named as ‘cnu-miR2673’ following the miRNA nomenclature procedure (Griffiths-Jones and Sam., 2006) .
The understanding of target gene function of miRNA is necessary step to know its regulation. The nature of conserved miRNAs in plants is an important factor that allows the finding of the target genes based on their complementarity or near complementarity of miRNAs to their respective targets( Zhang et al., 2006).We have found the five target genes for this one miRNA family based on our filtering criteria using the psRNAtarget tool using the available transcripts of Arabidopsis thaliana. All of the putative target genes seem to inhibit through cleavage mode by the identified miRNA and it has multiple targets as shown in Table 3 and Fig. 4.
| Table 3: List of the potential targets of newly identified miRNA in coconut | ||||||
| Target Acc. | e-Value | Inhibition | Target Desc. | GO Biological Process | GO Molecular Function | GO Cellular Component |
| AT3G25450.1 | 0 | Cleavage | Transposable element Copia-like retrotransposon family | Not Available | Not Available | Not Available |
| AT2G37650.1 | 0.5 | Cleavage | GRAS family transcription factor | | regulation of transcription, DNA-templated, transcription, DNA-templated | transcription factor activity, sequence-specific DNA binding | nucleus |
| AT1G10940.2 | 1 | Cleavage | Protein kinase superfamily, Encodes a plant protein kinase similar to the calcium/calmodulin-dependent protein kinase subfamily and the SNF1 kinase subfamily (SnRK2) whose activity is activated by ionic (salt) and non-ionic (mannitol) osmotic stress. | intracellular signal transduction, primary root development, protein phosphorylation, response to abscisic acid, response to osmotic stress, response to salt stress | ATP binding, kinase activity, protein binding, protein kinase activity, protein serine/threonine kinase activity | cytosol, membrane, nucleus |
| AT1G10940.1 | 1 | Cleavage | Protein kinase superfamily, Encodes a plant protein kinase similar to the calcium/calmodulin-dependent protein kinase subfamily and the SNF1 kinase subfamily (SnRK2) whose activity is activated by ionic (salt) and non-ionic (mannitol) osmotic stress. | intracellular signal transduction, primary root development, protein phosphorylation, response to abscisic acid, response to osmotic stress, response to salt stress | ATP binding, kinase activity, protein binding, protein kinase activity, protein serine/threonine kinase activity | cytosol, membrane, nucleus |
| AT5G06710.2 | 1 | Cleavage | homeobox from Arabidopsis thaliana / Homeobox-leucine zipper protein | regulation of transcription, DNA-templated, transcription, DNA-templated | sequence-specific DNA binding, transcription factor activity, sequence-specific DNA binding | nucleus |
| AT5G06710.1 | 1 | Cleavage | homeobox from Arabidopsis thaliana / Homeobox-leucine zipper protein | regulation of transcription, DNA-templated, transcription, DNA-templated | sequence-specific DNA binding, transcription factor activity, sequence-specific DNA binding | nucleus |
| AT1G21590.1 | 1.5 | Cleavage | Protein kinase protein with adenine nucleotide alpha hydrolases-like domain | Protein phosphorylation, response to stress | ATP binding, hydrolase activity, kinase activity, protein serine/threonine kinase activity | nucleus |
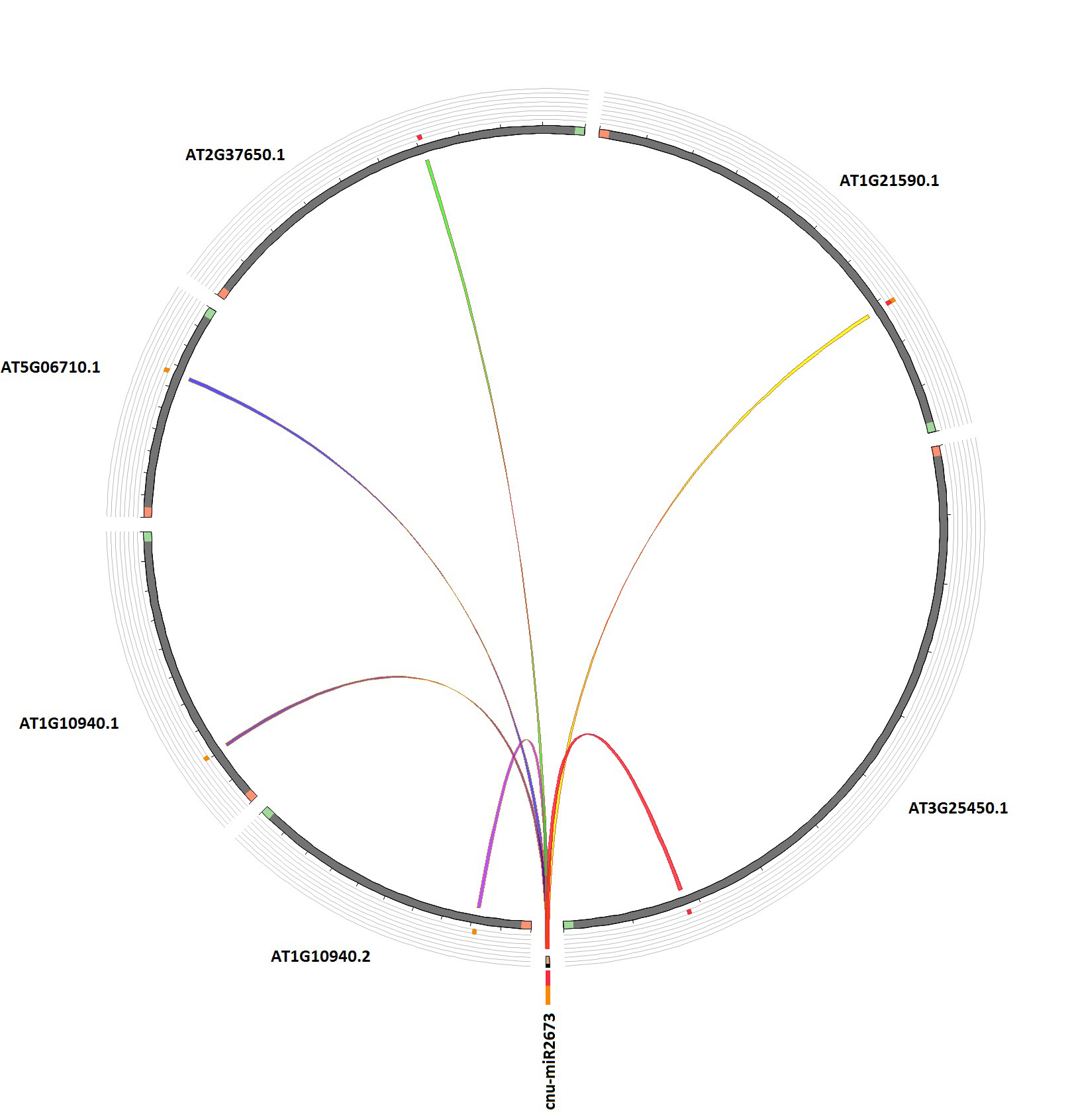
The predicted targets involves two transcription factors namely Homeobox leucine zipper family and GRAS family of transcription factor. HD-ZIP (Homeobox-leucine zipper protein) has homeodomain (HD) and linear zipper motif present. This class of transcription factors is unique to plants and are involved in plant growth and development especially in response to abscisic acid, abiotic stress, shade avoidance and auxin signaling (Ariel et al., 2007; Elhiti et al., 2009). GRAS proteins act as transcription factors, a number of which have nuclear localization signal for localization of several other proteins (Tian et al., 2004). It has important functions in GA (Gibberilic acid) and light signaling and regulation of root patterning (Hirsch et al., 2009) .
We have found one of the target gene for Copia retrotransposons. These retrotransposons are flanked by long terminal repeats (LTRs) that certain promoter and downstream controls elements their internal domain usually contains the genes required for reverse transcriptase, integrase and gag (Wilhelm and Wilhelm.,2001). The gene that had Copia like transposons has blast match to gag pol protein from Glycine max and previous findings support the existence of SIRI, a copia/Ty1-like retrotransposon element encoding a retroviral envelope like protein assumed to be originated from tomato genome through horizontal gene transfer (Cheng et al., 2009). Two of the target genes showed the exiatence of protein kinase out of which one encodes the calcium/calmodulin mediated kinase super subfamily and SNF1 kinase subfamily activated during ionic and non-ionic omotic stress. As exemplified, CDPKs (Calcium dependent protein kinases) play major roles in development for recycling transcription to hormone levels and are known to function in abiotic stress response and ABA signaling through phosphorylation activity(Ishida et al., 2008; Mori et al., 2006). On the other hand, SnRK(SNF kinases) are expressed in nucleus during seed development and germination in Arabidopsis. Any alteration in these protein induces gene expression change, both, in the up-regulation of ABA repressive genes and down-regulation of ABA inducible genes. The alteration of gene expression leads to loss of dormancy and growth defects during seed development (Nakashima et al., 2009). The other kinase protein with adenine alpha hydrolases phosphorylates proteins fuctions in response to stress with probable interaction with serine/threonine to ATP.
As in plants, for every 10,000 EST sequences is expected to contain one miRNA (Zhang et al., 2006). But, from our study, we propose that even 1000-2000 ESTs could be utilized to mine the conserved potential miRNAs across the non-model crops. The findings from this study indicate that coconut miRNA mir 2673 possibly targets both transcription factors and individual specific genes. Hence, the methodology adopted in this research will enumerate the understanding of regulatory miRNA in coconut in much rapid pace in near future.
Conclusion
The quest for potential miRNA is a highlighted research routine in the field of transcriptomics. However, over the recent years the miRNAs from non-model plants have been explored using ESTs. In this research, attempts has been made to screen miRNAs from 1008 ESTs reported in C.nucifera by employing, EST based homology search method, which resulted in one novel miRNA. Its putative function of gene targets were also catalogued. The impact of this study will be a starting point in understanding the miRNA biogenesis and its structure in Cocos nucifera and to explore repertoire of small RNAs that contribute to different biological mechanisms of the plant system. Since the genetic information is limited, the present in silico approach will help to improve the miRNAome in C.nucifera through further experimental validation.
Acknowledgement
The authors gratefully acknowledge the Bioinformatics Infrastructure Facility sponsored by Department of Biotechnology, Government of India, New Delhi. They also acknowledge the Head and Coordinator, Department of Biotechnology, University of Calicut, Kerala.
References
Akter, A., Islam, M.M., Mondal, S.I., Mahmud, Z., Jewel, N.A., Ferdous, S., Amin, M.R. and Rahman, M.M., 2014. Computational identification of miRNA and targets from expressed sequence tags of coffee (Coffea arabica). Saudi Journal of Biological Sciences, 21(1), pp.3-12.
Ariel, F.D., Manavella, P.A., Dezar, C.A. and Chan, R.L., 2007. The true story of the HD-Zip family. Trends in plant science, 12(9), pp.419-426.
Bartel, D.P., 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. cell, 116(2), pp.281-297.
Cheng, X., Zhang, D., Cheng, Z., Keller, B. and Ling, H.Q., 2009. A new family of Ty1-copia-like retrotransposons originated in the tomato genome by a recent horizontal transfer event. Genetics, 181(4), pp.1183-1193.
Cock, P.J., Chilton, J.M., Grüning, B., Johnson, J.E. and Soranzo, N., 2015. NCBI BLAST+ integrated into Galaxy. GigaScience, 4(1), p.1.
Dai, X. and Zhao, P.X., 2011. psRNATarget: a plant small RNA target analysis server. Nucleic acids research, 39(suppl 2), pp.W155-W159.
Darzentas, N., 2010. Circoletto: visualizing sequence similarity with Circos. Bioinformatics, 26(20).
Das, A. and Mondal, T.K., 2010. Computational identification of conserved microRNAs and their targets in tea (Camellia sinensis). American Journal of Plant Sciences, 1(02), p.77.
Das, A., Chaudhury, S., Kalita, M.C. and Mondal, T.K., 2015. In silico identification, characterization and expression analysis of miRNAs in Cannabis sativa L. Plant Gene, 2, pp.17-24.
Dehury, B., Panda, D., Sahu, J., Sahu, M., Sarma, K., Barooah, M., Sen, P. and Modi, M.K., 2013. In silico identification and characterization of conserved miRNAs and their target genes in sweet potato (Ipomoea batatas L.) Expressed Sequence Tags (ESTs). Plant signaling & behavior, 8(12), p.e26543.
Elhiti, M. and Stasolla, C., 2009. Structure and function of homodomain-leucine zipper (HD-Zip) proteins. Plant Signaling & Behavior, 4(2), pp.86-88.
Govaerts, R., 2003. World Checklist of Selected Plant Families Database in ACCESS: 1-216203. The Board of Trustees of the Royal Botanic Gardens, Kew.
Griffiths-Jones, S., 2006. miRBase: the microRNA sequence database. MicroRNA Protocols, pp.129-138.
Hirsch, S. and Oldroyd, G.E., 2009. GRAS-domain transcription factors that regulate plant development. Plant Signaling & Behavior, 4(8), pp.698-700.
Ishida, S., Yuasa, T., Nakata, M. and Takahashi, Y., 2008. A tobacco calcium-dependent protein kinase, CDPK1, regulates the transcription factor REPRESSION OF SHOOT GROWTH in response to gibberellins. The Plant Cell, 20(12), pp.3273-
3288.
Khraiwesh, B., Zhu, J.K. and Zhu, J., 2012. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms, 1819(2), pp.137-148.
Lelandais-Brière, C., Naya, L., Sallet, E., Calenge, F., Frugier, F., Hartmann, C., Gouzy, J. and Crespi, M., 2009. Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. The Plant Cell, 21(9), pp.2780-2796.
Li, W. and Godzik, A., 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22(13), pp.1658-1659.
Millar, A.A. and Gubler, F., 2005. The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. The Plant Cell, 17(3), pp.705-721.
Mori, I.C., Murata, Y., Yang, Y., Munemasa, S., Wang, Y.F., Andreoli, S., Tiriac, H., Alonso, J.M., Harper, J.F., Ecker, J.R. and Kwak, J.M., 2006. CDPKs CPK6 and CPK3 function in ABA regulation of guard cell S-type anion-and Ca 2+-permeable channels and stomatal closure. PLoS Biol, 4(10), p.e327.
Nakashima, K., Fujita, Y., Kanamori, N., Katagiri, T., Umezawa, T., Kidokoro, S., Maruyama, K., Yoshida, T., Ishiyama, K., Kobayashi, M. and Shinozaki, K., 2009. Three Arabidopsis SnRK2 protein kinases, SRK2D/SnRK2. 2, SRK2E/SnRK2. 6/OST1 and SRK2I/SnRK2. 3, involved in ABA signaling are essential for the control of seed development and dormancy. Plant and Cell Physiology, 50(7), pp.1345-1363.
Panda, D., Dehury, B., Sahu, J., Barooah, M., Sen, P. and Modi, M.K., 2014. Computational identification and characterization of conserved miRNAs and their target genes in garlic (Allium sativum L.) expressed sequence tags. Gene, 537(2), pp.333-342.
Reinhart, B.J., Weinstein, E.G., Rhoades, M.W., Bartel, B. and Bartel, D.P., 2002. MicroRNAs in plants. Genes & development, 16(13), pp.1616-1626.
Rhoades, M.W., Reinhart, B.J., Lim, L.P., Burge, C.B., Bartel, B. and Bartel, D.P., 2002. Prediction of plant microRNA targets. Cell, 110(4), pp.513-520.
Singh, J. and Nagaraju, J., 2008. In silico prediction and characterization of microRNAs from red flour beetle (Tribolium castaneum). Insect molecular biology, 17(4), pp.427-436.
Singh, N. and Sharma, A., 2014. In-silico identification of miRNAs and their regulating target functions in Ocimum basilicum. Gene, 552(2), pp.277-282.
Singh, N., Srivastava, S. and Sharma, A., 2016. Identification and analysis of miRNAs and their targets in ginger using bioinformatics approach. Gene, 575(2), pp.570-576.
Sunkar, R. and Jagadeeswaran, G., 2008. In silico identification of conserved microRNAs in large number of diverse plant species. BMC plant biology, 8(1), p.1.
Sunkar, R., Kapoor, A. and Zhu, J.K., 2006. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. The Plant Cell, 18(8), pp.2051-2065.
Tian, C., Wan, P., Sun, S., Li, J. and Chen, M., 2004. Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant molecular biology, 54(4), pp.519-532.
Wilhelm, M. and Wilhelm, F.X., 2001. Reverse transcription of retroviruses and LTR retrotransposons. Cellular and Molecular Life Sciences CMLS, 58(9), pp.1246-1262.
Yang, T., Xue, L. and An, L., 2007. Functional diversity of miRNA in plants. Plant Science, 172(3), pp.423-432.
Yin, Z., Li, C., Han, X. and Shen, F., 2008. Identification of conserved microRNAs and their target genes in tomato (Lycopersicon esculentum). Gene, 414(1), pp.60-66.
Zhang, B., Pan, X. and Anderson, T.A., 2006. Identification of 188 conserved maize microRNAs and their targets. Febs Letters, 580(15), pp. 3753-3762.
Zhang, B., Pan, X., Cannon, C.H., Cobb, G.P. and Anderson, T.A., 2006. Conservation and divergence of plant microRNA genes. The Plant Journal, 46(2), pp. 243-259.
Zhang, B., Pan, X., Cobb, G.P. and Anderson, T.A., 2006. Plant microRNA: a small regulatory molecule with big impact. Developmental biology, 289(1), pp. 3-16.
Zhang, B.H., Pan, X.P., Cox, S.B., Cobb, G.P. and Anderson, T.A., 2006. Evidence that miRNAs are different from other RNAs. Cellular and Molecular Life Sciences CMLS, 63(2), pp. 246-254.
Zhang, B.H., Pan, X.P., Wang, Q.L., George, P.C. and ANDERSON, T.A., 2005. Identification and characterization of new plant microRNAs using EST analysis. Cell research, 15(5), pp.336-360.
Zuker, M., 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research, 31(13), pp.3406-3415.

