Department of Biological Science Faculty of Science, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia
Corresponding author email: haniolfat@hotmail.com
Article Publishing History
Received: 18/11/2020
Accepted After Revision: 27/03/2021
BLM helicase protein plays important role in DNA replication and maintains the genomic integrity. Variation in BLM helicase gene resulted defect in DNA repair mechanism and are reported to be associated with bloom syndrome (BS) and cancer. Computational analysis of SNPs in BLM helicase gene has been performed to identify, characterize the pathogenic SNPs using bioinformatics approach. SNPs data has been obtained from dbSNP database for human BLM helicase (P54132). There were 1003 SNPs mapped to missense, 19890 SNPs mapped to intron, while 550 SNPs mapped to 5’UTR, 176 SNPs mapped to 3’UTR, 21551 mapped to total SNPs of different variation class and 11 SNPs mapped to pathogenic misense in human BLM helicase gene . 6 nsSNPs of 11 pathogenic missense are found to be deleterious or damaging by all four prediction tools. These 6 nsSNPs rs367543034 of mutation G952V, rs367543023 of mutation H963Y, rs137853153 of mutation C1036F, rs367543029 of mutation C1055Y, rs367543032 of mutation D1064G and rs367543025 of mutation C1066Y can be further investigated along with native protein. These mutations in BLM gene may have potential to be used as an important prognostic marker for detection of cancer, particularly for for surgically-treated lung adenocarcinoma, (Yang et al 2020).
nsSNP; Bloom Syndrome; In Silico Analysis; BLM
Ali H. M, Firoz A. Identification and Analysis of Pathogenic nsSNPs in Human Bloom Syndrome Helicase Bene BLM. Biosc.Biotech.Res.Comm. 2021;14(1).
Ali H. M, Firoz A. Identification and Analysis of Pathogenic nsSNPs in Human Bloom Syndrome Helicase Bene BLM. Biosc.Biotech.Res.Comm. 2021;14(1). Available from: <a href=”https://bit.ly/3rEdbYL”>https://bit.ly/3rEdbYL</a>
Copyright © Ali and Firoz This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) https://creativecommns.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
BLM gene encodes an important nuclear protein BLM helicase (Eladad et al., 2005), which involved in DNA replication and maintains the genomic integrity (Manthei et al., 2013). BLM is a 3’ to 5’ DNA helicase that belongs to conserved RecQ helicase family (Imamura et al., 2003). Helicases are very crucial for unwinding duplex DNA to produce the transient single-stranded DNA intermediates necessary for replication, recombination, and repair (Hall et al., 1999, Schmid et al., 1992). In a complex with topoisomerase Topo IIIa and Rmi1/Rmi2, BLM helicase repair DNA double-strand breaks through homologous recombination (HR) pathway (Matson et al., 1994). Consequently, cells lacking functional BLM show ~10-fold raising in chromatid breaks, and mitotic recombination, (Hickson et al., 2003). Bloom syndrome (BS) is a rare autosomal recessive genetic disorder caused by pathogenic variants in the BLM gene.
Symptoms of BS include low birth weight, dolichocephaly (long, narrow head), congenital short stature, growth retardation sun-sensitive facial rash, an elevated risk of diabetes mellitus, reduced fertility and immune deficiency (Shastri et al., 2015). Absence of BLM protein activity causes defect in DNA repair, increased rate of mutations and thus risk of cancer (Arora et al., 2014). BLM gene transcribes a 97.93 kb long precursor-mRNA having 21 exons, which code 1417 amino acid protein. Literature support that a large number of BS patients shows insertion, deletion and missense mutation that change the amino acid or nonsense mutations which introduce premature stop in the BLM gene and thus inactivate the BLM helicase (Ellis et al., 1995, Foucault et al., 1997, German et al., 2007 Mclaren et al 2016 Yang et al 2020).
Several articles have stated effectiveness in identifying the deleterious and disease associated mutations, thus predicting the pathogenic SNPs in correlation to their functional and structural damaging properties ( Adzhubei et al., 2010 Choi et al 2015). Computational studies have previously provided an efficient platform for evaluation and analysis of genetic mutations for their pathological consequences and in determining their underlying molecular mechanism. Single nucleotide polymorphism (SNPs) is a common genetic variation contributing greatly towards the phenotypic variations (Hecht et al., 2015). SNPs can alter the functional consequences of proteins. In the coding region of gene, SNPs may be synonymous, non-synonymous (nsSNPs) or nonsense. Synonymous SNPs changes the nucleotide base residue but does not change the amino acid residue in protein sequence due to degeneracy of genetic code.
The nsSNPs also called missense variants, alter amino acid residue in protein sequence and thus change the function of protein through altering protein activity, solubility and protein structure. (Calabrese et al., 2009). SNPs have been emerged as the genetic markers for many diseases and there are many SNPs markers available in the public databases. hundreds of new SNPs have been mapped to human BLM genes. However, not all SNPs are functionally important. Despite extensive studies of helicase proteins in human and effect of their polymorphism in cancer (Hecht et al., 2015), no attempt was made to analyze to establish the functional consequences of pathogenic nsSNPs of BLM gene. The aim of this study is to identify the high pathogenic SNPs of BLM gene and determine functional consequences using computational methods.
MATERIAL AND METHODS
SNPs Dataset : The SNPs of the BLM helicase gene (Uniport id P54132) were retrieved from the dbSNP database (Sherry et al., 1999). Keyword “Human BLM” used as our search term. Furthermore, it is filtered by selecting variation class as SNV, function class as missense, clinical significance as pathogenic.
Predicting deleterious and damaging nsSNPs:In order to predict the damaging or deleterious nsSNPs, multiple consensus tools were employed by using online tool VEP (http://www.ensembl.org/Tools/VEP). VEP advantages include: it uses latest human genome assembly GRCh38.p10, and can predict thousands of SNPs from multiple tools including SIFT, PROVEAN, Condel, and PolyPhen-2, at a time (McLaren et al., 2016). 11 nsSNP rs-ids were uploaded to VEP tool to get the prediction results
Sift: The algorithm predicted that the tolerant and intolerant coding base substitution based upon properties of amino acids and homology of sequence (Ng PC et al., 2003). The tool considered that vital positions in the protein sequence have been conserved throughout evolution and therefore substitutions at conserved alignment position is expected to be less tolerated and affect protein function than those at diverse positions. SIFT predicted substituted amino acid as damaging at default threshold score <0.05, while score ³ 0.05 is predicted as tolerated.
Provean: The online tool uses an alignment-based scoring method for predicting the functional consequences of single and multiple amino acid substitutions, and in-frame deletions and insertions (Choi et al., 2012). The tool has a default threshold score, i.e. -2.5, below which a protein variant is predicted as deleterious, and above that threshold, a protein variant is neutral.
Condel (CONsensusDELeteriousness): This tool evaluates the probability of missense single nucleotide variants (SNVs) deleterious. it computes a weighted average of the scores of SIFT, PolyPhen2, MutationAssessor and FatHMM (González-Pérez et al., 2011).
Polyphen-2: This tool is predicting the structural and functional consequences of a particular amino acid substitution in human protein (Ramensky et al., 2002). Prediction of PolyPhen-2 server [20] is based on a number of features including information of structural and sequence comparison. The PolyPhen-2 score varies between 0.0 (benign) to 10.0 (damaging). The PolyPhen-2 prediction output categorizes the SNPs into three basic categories, benign (score < 0.2), possibly damaging, (score between 0.2 and0.96), or probably damaging (score >0.96).
RESULTS AND DISCUSSION
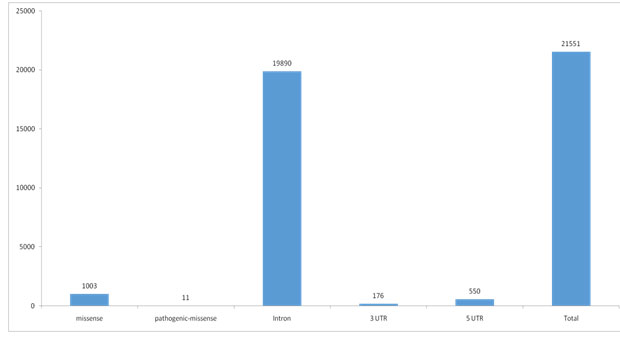
11 rs-ids of pathogenic nsSNPs mapped in human BLM helicase gene was downloaded from dbSNP database of NCBI (Table 1), after filtering variation class SNV, function class missense and clinical significance as pathogenic, there were 1003 SNP mapped to missense, 19890 SNPs mapped to intron, while 550 SNPs mapped to 5’UTR, 176 SNPs mapped to 3’UTR and 21551 mapped to total SNPs of different variation class (Figure 1). Some rsIDs are associated with multiple SNPs and therefore fall in different classes.
Figure 1: Number of SNPs in different function class of BLM helicase gene of human from dbSNP database showing missense (1003), pathogenic missense (11), intron (19890), 3UTR (176), 5UTR (550) and Total (21551).
Predicting deleterious and damaging pathogenic nsSNPs: In order to predict the damaging or deleterious pathogenic nsSNPs multiple consensus tools were employed. Initially, online tool VEP was used . VEP advantages include: it uses latest human genome assembly GRCh38.p10, and can predict thousands of SNPs from multiple tools including SIFT, Condel, and PolyPhen-2, at a time. 11 nsSNP accession numbers were uploaded to VEP tool and the prediction results were taken on default scores of consensus tools based on sequence and structure homology methods: (a) SIFT (score <-0.5) (b) Polyphen (score >0.96) (c) PROVEAN (score< 2.5) and Condel (score >0.522). In order to get a very high confident nsSNPs impacting structure and function of BLM gene, 6 nsSNPs (Table 1) are found to be deleterious or damaging by all four prediction tools. These 6 nsSNPs rs367543034 of mutation G952V, rs367543023 of mutation H963Y, rs137853153 of mutation C1036F, rs367543029 of mutation C1055Y, rs367543032 of mutation D1064G and rs367543025 of mutation C1066Y.
Table.1 Prediction of 11 pathogenic missense SNPs of BLM helicase gene using prediction tools such as SIFT, Condel, Polyphen and PROVEAN, deleterious predicted by all four tools are shown in bold.
| SNP-ids | AA-Change | SIFT | PolyPhen | PROVEAN | Condel |
| rs746195311 | E69K | deleterious | benign | neutral | neutral |
| rs367543030 | S104L | deleterious | Possibly damaging | neutral | neutral |
| rs1477193473 | E488K | tolerated | benign | neutral | neutral |
| rs200389141 | Q548K | deleterious | benign | neutral | neutral |
| rs367543034 | G952V | deleterious | Probably damaging | deleterious | deleterious |
| rs367543023 | H963Y | deleterious | probably
damaging |
deleterious | deleterious |
| rs137853153 | C1036F | deleterious | Probably damaging | deleterious | deleterious |
| rs367543029 | C1055Y | deleterious | probably damaging | deleterious | deleterious |
| rs367543032 | D1064G | deleterious | probably damaging | deleterious | deleterious |
| rs367543025 | C1066Y | deleterious | probably damaging | deleterious | deleterious |
| rs367543017 | S1093L | deleterious | Possibly damaging | deleterious | Neutral |
This analysis shows that six SNPs, G952V, H963Y, C1036F, C1055Y, D1064G and C1066Y have high prevalence for disease association of BLM, the mutation in cysteines (C1036F, C1055Y, C1066Y) and glutamate (D1064V) are in the Zn binding subdomain, which results in the loss of Zn binding upon mutation and alters the function of BLM helicase is reported (Guo et al., 2005). These mutation in RQC domain affect the highly conserved cysteine residues involved in Zn coordination . While mutation in Glycine G952V and mutation in histidine H963Y which alter amino acid residues in the ATPase domain also reported involved in cellular defects (Shastri and Schmidt 2015).
CONCLUSION
This computational analysis of SNPs of the human BLM protein identified 6 highly damaging pathogenic nsSNPs. Prediction analysis shows that SNPs G952V, H963Y, C1036F, C1055Y, D1064G and C1066Y have high prevalence for disease association. Data implies that the reported nsSNPs could potentially alter structure and hence the function of BLM protein resulting in pathogenicity with abnormal symptoms describing the disease states. These nsSNPs associated with significant pathogenicity will offer valuable information in selecting SNPs that are expected to have impending functional influence and contribute in understanding the functional roles of this gene.
ACKNOWLEDGEMENTS
This work was not supported by any grants agency. We acknowledge with thanks Deanship of Scientific Research (DSR), at King Abdulaziz University, Jeddah, KSA for providing their support.
REFERENCES
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. (2010) A method and server for predicting damaging missense mutations. Nat Methods. 7(4) pp 248-9
Arora H, Chacon A H, Choudhary S, McLeod M P, Meshkov L, Nouri K, amd Izakovic J. (2014) Bloom syndrome. International journal of dermatology 53(7) pp 798–802.
Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R.(2009) Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 30(8):1237-44.
Choi Y, Chan AP. (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 15;31(16) pp 2745-7.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 7(10): e46688.
Eladad S, Ye TZ, Hu P, Leversha M, Beresten S, Matunis MJ and Ellis NA (2005) Intra-nuclear trafficking of the BLM helicase to DNA damage-induced foci is regulated by SUMO modification, Human Molecular Genetics, Vol 14, Issue 10, pp 1351–1365
Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. (1995) The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 17;83(4):655-66.
Foucault F, Vaury C, Barakat A, Thibout D, Planchon P, Jaulin C, Praz F, Amor-Guéret M. (1997) Characterization of a new BLM mutation associated with a topoisomerase II alpha defect in a patient with Bloom’s syndrome. Hum Mol Genet. 6(9) pp 1427-34.
German J, Sanz MM, Ciocci S, Ye TZ, Ellis NA.(2007) Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 28(8) pp743-53.
González-Pérez A, López-Bigas N.(2011) Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 8;88(4):440-9.
Guo R, Rigolet P, Zargarian L, Fermandjian S, Xi XG, (2005) Structural and functional characterizations reveal the importance of a zinc binding domain in Bloom’s syndrome helicase, Nucleic Acids Research, Volume 33, pp 3109–3124.
Hall MC and Matson SW, (1999) Helicase motifs: the engine that powers DNA unwinding. Mol Microbiol, 34(5) pp 867-77.
Hecht M, Bromberg Y and Rost B.(2015) Better prediction of functional effects for sequence variants. BMC Genomics 16, S1 1186/1471-2164-16-S8-S1
Hickson I.D, (2003) RecQ helicases: caretakers of the genome. Nat Rev Cancer, 3(3) pp. 169-78.
Imamura O, Campbell JL (2003) The human Bloom syndrome gene suppresses the DNA replication and repair defects of yeast dna2 mutants, Proceedings of the National Academy of Sciences, Vol 100 (14) pp 8193-8198
Manthei KA, Keck JL, (2013) The BLM dissolvasome in DNA replication and repair, Cell Mol Life Sci, 70(21) pp 4067-4084
Matson SW, DW Bean, and J.W. George, (1994) DNA helicases: enzymes with essential roles in all aspects of DNA metabolism. Bioessays, 16(1) pp 13-22.
McLaren W, Gil L, Hunt SE.(2016) The Ensembl Variant Effect Predictor. Genome Biol 17, 122
Ng PC, Henikoff S. (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 1;31(13):3812-4.
Ramensky V, Bork P, Sunyaev S. (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids ResSep 1;30(17):3894-900.
Schmid SR and P Linder, (1992) D-E-A-D protein family of putative RNA helicases. Mol Microbiol, 6(3) pp. 283-91.
Shastri VM, Schmidt KH (2015) Cellular defects caused by hypomorphic variants of the Bloom syndrome helicase gene BLM, Mol Genet Genomic Med, 4(1), 106-119.
Sherry ST, Ward M, Sirotkin K.(1999) dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 9(8):677-9.
Yang X , Guohui Wang , Runchuan Gu , Xiaohong Xu and Guangying Zhu (2020) A signature of tumor DNA repair genes associated with the prognosis of surgically-resected lung adenocarcinoma Peer J Published November 26, 2020 PubMed ID 33304656 DOI 10.7717/peerj.10418

