Department cum National Centre for Human Genome Studies & Research, Panjab University, Chandigarh, India
Corresponding author email: ranvir1@pu.ac.in
Article Publishing History
Received: 28/10/2020
Accepted After Revision: 07/12/2020
Bacterial pathogens have developed multidrug resistance which is a major challenge to global health. Fusidic acid (FA) is a steroid antibiotic commonly used against gram positive Staphylococcus aureus (S. aureus) infections in clinics. It binds to Elongation Factor-G (EF-G) and inhibits translation by stalling EF-G on ribosome after GTP hydrolysis. Disease causing infectious gram positive bacteria such as Mycobacterium tuberculosis (M. tuberculosis), Mycobacterium leprae (M.leprae) and Bacillus anthracis (B. anthracis) pose major health challenge. In order to investigate if FA can be repurposed against these bacterial pathogens, we carried out multiple sequence alignment of EF-G from these bacterial species followed by prediction of their three dimensional structure using homology modeling. These predicted dimensional structures after validation were used for structural analysis with crystal structure of EF-G from Thermus thermophilus (T.thermophilus) and S.aureus.
Molecular docking was performed to dock FA into its putative binding site of the predicted three dimensional structures of these bacterial EF-G.Multiple sequence alignment of EF-G sequences from these bacteria showed sufficient sequence identity. Homology models of EF-G from these bacteria were compared with available crystal structure of EF-G from Thermus thermophilus (T.thermophilus) which revealed overall conserved tertiary structure. Docking of FA to homology models suggested that the antibiotic in principle bind to EF-G from these gram positive infectious bacteria. We therefore propose that fusidic acid as a strong potential drug candidate against infections caused by these gram positive bacteria.
Fusidic acid, Elongation Factor–G (EF-G), Gram Positive Bacteria, Infectious Diseases
Kapoor P, Singh R. Fusidic Acid as a Potential Drug Candidate Against Infectious Gram Positive Bacteria. Biosc.Biotech.Res.Comm. 2020;13(4).
Kapoor P, Singh R. Fusidic Acid as a Potential Drug Candidate Against Infectious Gram Positive Bacteria. Methods of Deodorizing Round scad (Decapterus maruadsi) for the Production of Protein Hydrolysates. Biosc.Biotech.Res.Comm. 2020;13(4). Available from: https://bit.ly/33k34yD
Copyright © Kapoor and Singh This is an Open Access Article distributed under the Terms of the Creative Commons Attribution License (CC-BY) https://creativecommons.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
Fusidic acid (FA) is a steroid antibiotic derived from fungus Fusidium coccineum and is commonly used against infections caused by gram positive bacterium Staphylococcus aureus (S.aureus) (Verbist, 1990). It has an unusual spectrum of activity that includes corynebacteria, nocardia, anaerobes, and Neisseria species, but is used almost exclusively as an anti-staphylococcal agent (Collignon, and Turnidge,1999.). FA inhibits protein synthesis by acting directly on elongation factor-G (EF-G) (Bodley et al., 1969). EF-G is a GTPase which catalyzes the translocation step during elongation phase of protein synthesis. EF-G in complex with GTP translocates the peptidyl-tRNA from A-site to P-site along with GTP hydrolysis. After GTP hydrolysis, EF-G•GDP dissociates from ribosome (Rodnina et al.1999). In the presence of fusidic acid, EF-G remains bound to the ribosome after GTP hydrolysis, sterically blocking the next stage in protein synthesis. (Burns et al., 1974; , Willi et al., 1975 Belardinelli and Rodnina, 2017).
Resistance to fusidic acid is primarily caused by mutations in fusA gene which encodes EF-G and several mutants conferring FA resistance in EF-G from Salmonella typhimurium [S. typhimurium) and from S. aureus were identified and phenotypically characterized in vivo (Johanson and Hughes, 1994; Laurberg et al.,2000).. Crystal structures of EF-G have been determined from Thermus thermophilus (T. thermophilus) and S.aureus ( Ævarsson,et al.,1994; Chen et al., 2010; Czworkowski et al.,1994; Laurberg et al., 2000;). The protein is an elongated molecule composed of five domains (I-V) whose overall shape resembles that of a tadpole. Domain I (G domain) is the nucleotide binding domain. Domain I and II constitute a single globular unit while domain III, IV and V constitute another block. The domain interface (domain G/ III/ V) has been proposed to undergo conformational change implicating conformational dynamics of the molecule (Lin et al., 2015; Stark et al.,2000 Waterhouse et al 2018).
Availability of the structure of EF-G allowed to map FA resistant mutants as well as to provide possible explanation for their mechanism of action and for the probable binding site of FA. Main conclusions were a) resistant mutants are spread all over the EF-G indicating that few mutants might be directly interacting with FA binding site. b) Three prominent clusters of mutants were identified mapping to G domain, domain III and domain V. c) Most likely these mutations in clusters shall restrict the conformational dynamics of EF-G required for its function or shall modulate affinity of EF-G for ribosome/FA or both. d).The domain interface is the most likely binding site for fusidic acid. Among bacterial pathogens, M.leprae, M. tuberculosis and B.anthrasis have posed serious health challenges globally, (Katale et al 2020).
In this report, we have addressed the following issues: a) To what extent EF-G from these select bacteria display sequence identity to EF-G from T. thermophilus and S.aureus so as to permit homology modeling. b) Whether the tertiary structure is conserved among EF-G homology models when compared with EF-G crystal structure and c) If fusidic acid can bind to proposed putative binding site in these modeled structures. We report that there exists significant overall sequence identity among EF-G sequences from these bacteria to allow homology modeling. The overall structural similarity of these models is quite good when compared with EF-G structure from T. thermophilusand S.aureus. The domain interface, the putative binding site for fusidic acid is quite conserved at sequence as well at structure level. Docking of fusidic acid to these models demonstrated that domain interface is the only cavity, where fusidic acid may bind. These results strongly suggest that fusidic acid as a potential drug candidate against these pathogenic bacteria.
MATERIAL AND METHODS
Sequence alignment: EF-G sequences of select infectious gram positive bacteria were retrieved from the SWISS PROT (Table 1.). Multiple sequence alignment of the sequences was performed using CLUSTALW with default parameters (Larkin et al., 2007). Homology modeling: The three dimensional structure of the EF-G from the select bacteria were modeled using the SWISS MODEL (Waterhouse et al., 2018). Stereochemical quality of the homology models were validated through PROCHECK (Laskowski et al.,2001) Visualization of the models and analysis was performed using PyMOL and figures were also generated through PyMOL. Molecular Docking: Fusidic acid was docked with homology models using Patchdock (Schneidman-Duhovny et al., 2005). Patchdock is geometry- based molecular docking algorithm which evaluates molecular shape complementarity between macromolecule and ligand.
RESULTS AND DISCUSSION
Elongation factor-G belongs to the GTPase superfamily of proteins. The members of this superfamily shares a nucleotide binding domain and two structural dynamic regions, called switch I, switch II, along with phosphate – binding loop (P- loop) which forms the part of nucleotide binding site (Bourne et al., 1991). The protein is composed of five domains and can be thought of two structural blocks, block 1(domain I and II) and block II (domains III – V). Block II can rotate with regard to block I (Lin et al., 2015). Alignment of EF-G sequences from these select gram positive bacteria reveals significant conservation of consensus elements of GTP binding proteins like P-loop (19-25), switch-I (40-65), switch II (84 -100). Also, secondary structural elements which are part of interface between domain G, domain III and domain V, like helix BG [91-100), helix CG (117-123) and helix BIII (456-467) are also quite conserved (Fig. 1).
Figure 1
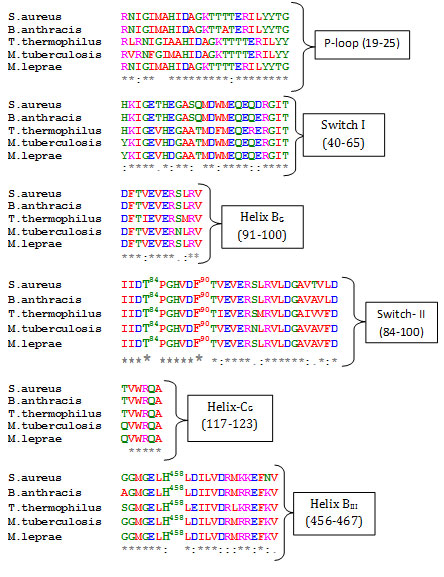
Fig1. Multiple Sequence Alignment of EF-G sequences. Identical residues are marked (*) and similar with (:). Bold and numbered residues shows few select residues of domain interface. Pairwise alignment of each of the EF-G sequence from these bacteria with that of T. thermophilus reveals significant overall sequence identity, 62 % with S. aureus, 61% with M. tuberculosis, 67% with B. anthracis and 60% with M. leprae (data not shown). Significant sequence overall identity of these bacterial sequences with EF-G from T. thermophilus and S. aureus allowed structure prediction of these bacterial EF-G using homology modeling. All the models were evaluated for their stereo-chemical quality. The homology models displayed satisfactory Ramachandran plot statistics (data not shown).The overall fold of the modeled structures are very similar to the elongated shape of the EF-G crystal structure (Fig. 2).
Figure 2
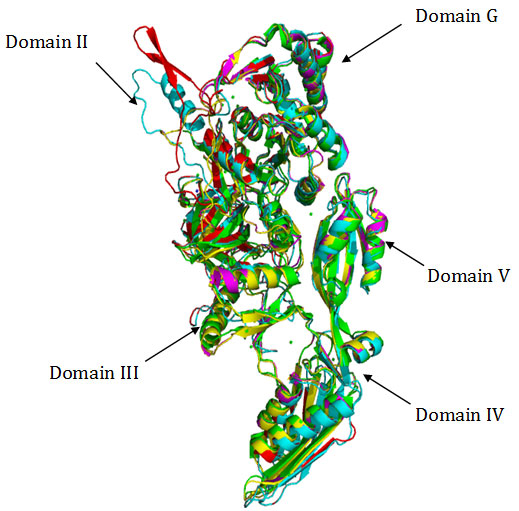
Fig2. Superimposition of Cα traces of crystal structure of EF-G from T. thermophilus (green), S. aureus (magenta) with homology models of EF-G from B.anthracis (yellow), , M. tuberculosis (cyan) and M.leprae (red). Superimposition of Cα backbone coordinates of EF-G from T. thermophilus with homology models showed root – mean – square deviations (rmsd’s) of 1.24 Å in case of M. tuberculosis, 1.29 in case of M. leprae and 0.72 Å in case of B. anthracis respectively. The crystal structure of EF-G from T. thermpophilus when superimposed with crystal structure of S.aureus (PDB 2xex) showed rmsd of 0.71 A.This reflects quite conservation of the overall fold of the protein in case of EF-G from these gram positive bacteria suggesting strong conservation of the tertiary structure.
It has been proposed that the probable binding site of fusidic acid is domain interface (Belardinelli and Rodnina 2017). Some of the key residues located at this interface are Thr84, Phe90, His458, Asp435, Thr437, G453, which are highly conserved in all the sequences (Fig.1).A close inspection of the domain interface of the models revealed, the putative binding pocket is also significantly conserved at structural level. The important elements of this interface are helix BG and helix CG from G domain, helix BIII and strand 4V from domain III and domain V respectively (Fig.3).
Figure 3
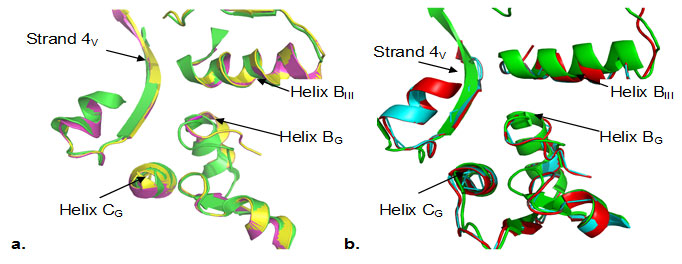
Fig3. Zoom in view of the superimposition of the backbone coordinates of domain interface (domain G/III/V) a). T.thermophilus (green) with B.anthracis (yellow), and S.aureus (magenta) b) T.thermophilus (green) with M.tuberculosis (cyan) and M.leprae (red). Secondary structure elements, helix BG, helix CG, helix BIII and strand4V are labeled. Spontaneous mutations observed under fusidic acid selective pressure are mainly clustered in helix BG and helix CG. The domain interface is structurally well conserved both in case of B. anthracis and S. aureus (Fig. 3a) as well as in case of M. tuberculosis and M. leprae (Fig.3b). Many of the domain interface residues (Phe90, His458, Asp435, Thr437, G453) in fact are known to confer strong resistance to fusidic acid upon mutation (Hansson et al., 2005). Structural aspects of fusidic acid resistance and sensitivity have emphasized the role of domain G/domain III/domain V interface as a key component of the FA binding site, (Hansson et al., 2005).
In order to investigate the domain interface as most probable binding cavity for fusidic acid, docking of fusidic acid with all homology models as well as with the crystal structure of EF-G from T. thermophilus and S.aureus was performed with Patchdock, a geometry based molecular docking algorithm. This algorithm focus on local shape feature matching. The sequential three major stages in the algorithm are a) molecular shape representation of the target and the ligand b) surface patch matching of the target and ligand c) Filtering and scoring of candidate complexes from second stage. Solutions with significant unacceptable steric clashes between target and ligand atoms are filtered out and discarded. Solutions with minor clashes (reflects conformational change of the molecular surface upon docking of the ligand) are scored based on geometric compatibility based on the size of the computed interface.
Thus solutions are ranked based on geometry scores, interface area size and desolvation. The top twenty solutions were analyzed and the best solution among them based on above mentioned scores as well as visual inspection was selected in each case. In order to evaluate the binding position, domain interface of each EF-G model was compared with the docked structure of crystal structure of EF-G from T. thermophilus by superimposition of protein Cα backbone coordinates. Inspection of the domain interface of docked structures of EF-G from T. thermophilus, along with S.aureus and B. anthracis, showed quite similar position and orientation of fusidic acid in the putative binding site(Fig.4a).
Figure 4
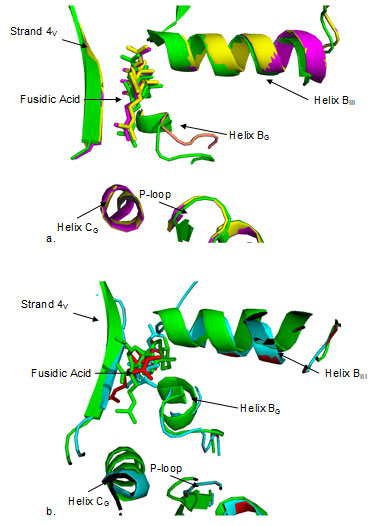
Fig4. Close up view of domain interface of EF-G structures docked with fusidic acid. Docked FA is shown in the same color as the color code of the protein. a) T. thermophilus (green) with B.anthracis (yellow), S.aureus (magenta) b) T. thermophilus with M.tuberculosis (cyan) and M.leprae (red). FA is docked in quite similar position as well as orientation in a) while the position seems to be similar in b), orientation is different in each case. Both in a) and b), FA is in close proximity to helix BG, helix BIII and strand 4V. Figure 4b shows the results of superimposition at domain interface when docked structure of EF-G from T. thermophilus is compared with that of M. tuberculosis and M. leprae. FA seems to bind almost in similar position in the interface as in case with S.aureus and B. anthracis (Fig. 4a), yet the orientation seems to be quite different. It is known that FA binds to EF-G only when it is bound to ribosome (Belardinelli and Rodnina 2017) .
Fusidic acid does not inhibits GTP hydrolysis and translocation by EF-G but prevents the dissociation of EF-G• GDP from ribosome binding (Borg et al., 2015). Nonetheless, structural mapping of fusidic acid resistant mutants in S. aureus and S. typhimurium led to proposal of a putative binding site for fusidic acid, implicating the interface of domain G with domain II and domain V as the most probable binding site (Laurbert et al., 2000) The current hypothesis is that that the domain interface is the most likely binding site for fusidic acid, especially implicating the role of Phe 90 in binding of FA to its putative site (Koripella et al., 2012). It is also interesing to note that some strong FA resistant mutations are located in the domain interface (Nagaev et al., 2001). Among these residues, Thr 84, Phe 90 are part of a loop which comprises switch II preceding helix BG . His 458 is part of helix BIII and G453 is part of domain interface.
Fusidic acid has been shown to be effective in vitro against certain clinical isolates of M. tuberculosis [Hoffner et al., 1990; Cicek-Saydam et al., 2001). Also strains of M. tuberculosis mono- or multiresistant to standard antituberculosis drugs have been reported to be susceptible to fusidic acid (Fuursted et al., 1992). It has been demonstrated that FA has excellent pharmacokinetics and in vitro activity against extracellular and intracellular M. leprae (Franzblau et al., 1992). There is only one reported clinical trial of FA for lepromatous leprosy [Franzblau et al., 1994). Little FA resistance has been reported against B. anthracis isolates [Odendaal et al., 1991). The results presented in this paper demonstrate significant conservation of EF-G sequence as well as structure along with strong conservation of domain interface among these gram positive pathogenic bacteria. These findings strongly suggests that FA shall not be exclusively used as anti-staphylococcal and has a potential to be used as a drug against these gram positive bacterial pathogens.
CONCLUSION
In conclusion, this study demonstrates that fusidic acid, an important antibiotic primary used against S.aureus can be repurposed for other gram positive infectious bacteria namely B. anthracis, M.leprae, and M.tuberculosis.
ACKNOWLEDGMENTS
Prashant Kapoor acknowledges Dept.cum NCHGSR, Panjab University for providing opportunity to carry out this work during his M.Sc dissertation thesis.
Conflict of interest: The authors declare no conflict of interest.
Contributions: RS conceived the work and PK carried out the work under guidance of RS. RS and PK analyzed the results. PK and RS wrote the manuscript. RS finalized the manuscript.
One Health approach in the prevention and control of mycobacterial infections in Tanzania: lessons learnt and future perspectives
REFERENCES
AEvarsson, A., Brazhnikov, E., Garber, M., Zheltonosova, J., Chirgadze, Y., al-Karadaghi, S., Svensson, L. and Liljas, A., 1994. Three-dimensional structure of the ribosomal translocase: elongation factor G from Thermus thermophilus. The EMBO Journal, 13(16), pp.3669-3677.
Belardinelli, R. and Rodnina, M., 2017. Effect of Fusidic Acid on the Kinetics of Molecular Motions During EF-G-Induced Translocation on the Ribosome. Scientific Reports, 7(1).
Bodley, J., Zieve, F., Lin, L. and Zieve, S., 1969. Formation of the ribosome-G factor-GDP complex in the presence of fusidic acid. Biochemical and Biophysical Research Communications, 37(3), pp.437-443.
Borg, A., Holm, M., Shiroyama, I., Hauryliuk, V., Pavlov, M., Sanyal, S. and Ehrenberg, M., 2014. Fusidic Acid Targets Elongation Factor G in Several Stages of Translocation on the Bacterial Ribosome. Journal of Biological Chemistry, 290(6), pp.3440-3454.
Bourne, H., Sanders, D. and McCormick, F., 1991. The GTPase superfamily: conserved structure and molecular mechanism. Nature, 349(6305), pp.117-127.
Burns, K. and Cannon, M., 1974. A resolution of conflicting reports concerning the mode of action of fusidic acid. FEBS Letters, 40(1), pp.219-223.
Chen, Y., Koripella, R., Sanyal, S. and Selmer, M., 2010. Staphylococcus aureus elongation factor G – structure and analysis of a target for fusidic acid. FEBS Journal, 277(18), pp.3789-3803.
Cicek-Saydam, C., Cavusoglu, C., Burhanoglu, D., Hilmioglu, S., Ozkalay, N. and Bilgic, A., 2001. In vitro susceptibility of Mycobacterium tuberculosis to fusidic acid. Clinical Microbiology and Infection, 7(12), pp.700-702.
Collignon, P. and Turnidge, J., 1999. Fusidic acid in vitro activity. International Journal of Antimicrobial Agents, 12, pp.S45-S58.
Czworkowski, J., Wang, J., Steitz, T. and Moore, P., 1994. The crystal structure of elongation factor G complexed with GDP, at 2.7 A resolution. The EMBO Journal, 13(16), pp.3661-3668.
Franzblau, S., Biswas, A. and Harris, E., 1992. Fusidic acid is highly active against extracellular and intracellular Mycobacterium leprae. Antimicrobial Agents and Chemotherapy, 36(1), pp.92-94.
Franzblau, S., Chan, G., Garcia-Ignacio, B., Chavez, V., Livelo, J., Jimenez, C., Parrilla, M., Calvo, R., Williams, D. and Gillis, T., 1994. Clinical trial of fusidic acid for lepromatous leprosy. Antimicrobial Agents and Chemotherapy, 38(7), pp.1651-1654.
Fuursted, K., Askgaard, D. and Faber, V., 1992. Susceptibility of strains of the Mycobacterium tuberculosis complex to fusidic acid. APMIS, 100(7-12), pp.663-667.
Hansson, S., Singh, R., Gudkov, A., Liljas, A. and Logan, D., 2005. Structural Insights into Fusidic Acid Resistance and Sensitivity in EF-G. Journal of Molecular Biology, 348(4), pp.939-949.
Hoffner, S., Olsson-Liljequist, B., Rydgård, K., Svenson, S. and Källenius, G., 1990. Susceptibility of Mycobacteria to fusidic acid. European Journal of Clinical Microbiology & Infectious Diseases, 9(4), pp.294-297.
Johanson, U. and Hughes, D., 1994. Fusidic acid-resistant mutants define three regions in elongation factor G of Salmonella typhimurium. Gene, 143(1), pp.55-59.
Johanson, U., Ævarsson, A., Liljas, A. and Hughes, D., 1996. The Dynamic Structure of EF-G Studied by Fusidic Acid Resistance and Internal Revertants. Journal of Molecular Biology, 258(3), pp.420-432.
Katale BZ, E Mbugi J Keyyu (2020) One Health approach in the prevention and control of mycobacterial infections in Tanzania: lessons learnt and future perspectives One Health Outlook 1(1):2 DOI: 10.1186/s42522-019-0002-1
Koripella, R., Chen, Y., Peisker, K., Koh, C., Selmer, M. and Sanyal, S., 2012. Mechanism of Elongation Factor-G-mediated Fusidic Acid Resistance and Fitness Compensation in Staphylococcus aureus. Journal of Biological Chemistry, 287(36), pp.30257-30267.
Larkin, M., Blackshields, G., Brown, N., Chenna, R., McGettigan, P., McWilliam, H., Valentin, F., Wallace, I., Wilm, A., Lopez, R., Thompson, J., Gibson, T. and Higgins, D., 2007. Clustal W and Clustal X version 2.0. Bioinformatics, 23(21), pp.2947-2948.
Laskowski, R., MacArthur, M., Moss, D. and Thornton, J., 1993. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26(2), pp.283-291.
Laurberg, M., Kristensen, O., Martemyanov, K., Gudkov, A., Nagaev, I., Hughes, D. and Liljas, A., 2000. Structure of a mutant EF-G reveals domain III and possibly the fusidic acid binding site 1 1Edited by I. A. Wilson. Journal of Molecular Biology, 303(4), pp.593-603.
Lin, J., Gagnon, M., Bulkley, D. and Steitz, T., 2015. Conformational Changes of Elongation Factor G on the Ribosome during tRNA Translocation. Cell, 160(1-2), pp.219-227.
Nagaev, I., Bjorkman, J., Andersson, D. and Hughes, D., 2001. Biological cost and compensatory evolution in fusidic acid-resistant Staphylococcus aureus. Molecular Microbiology, 40(2), pp.433-439.
Odendaal MW, Pieterson PM, de Vos V, Botha AD.1991. The antibiotic sensitivity patterns of Bacillus anthracis isolated from the Kruger National Park. Onderstepoort J Vet Res. 58(1):17-9.
Rodnina, M., Savelsbergh, A. and Wintermeyer, W., 1999. Dynamics of translation on the ribosome: molecular mechanics of translocation. FEMS Microbiology Reviews, 23(3), pp.317-333.
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R. and Wolfson, H., 2005. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Research, 33(Web Server), pp.W363-W367.
Stark, H., Rodnina, M., Wieden, H., van Heel, M. and Wintermeyer, W., 2000. Large-Scale Movement of Elongation Factor G and Extensive Conformational Change of the Ribosome during Translocation. Cell, 100(3), pp.301-309.
Verbist, L., 1990. The antimicrobial activity of fusidic acid. Journal of Antimicrobial Chemotherapy, 25(suppl B), pp.1-5.
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F., de Beer, T., Rempfer, C., Bordoli, L., Lepore, R. and Schwede, T., 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), pp.W296-W303.
Willie, G., Richman, N., Godtfredsen, W. and Bodley, J., 1975. Translocation. XV. Characteristics of and structural requirements for the interaction of 24,25-dihydrofusidic acid with ribosome.elongation factor G complexes. Biochemistry, 14(8), pp.1713-1718.

