1Genetics and Agricultural Biotechnology Institute of Tabarestan (GABIT), Sari Agricultural Sciences and Natural Resources University, PO Box 578, Sari, Iran
2National Institute of Genetics Engineering and Biotechnology (NIGEB), Tehran, Iran
3Department of Plant Breeding and Biotechnology, Gorgan University of Agricultural Sciences & Natural Resources, Gorgan, Iran
4RG Abiotic Stress Genomics, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben, Germany
Article Publishing History
Received: 01/08/2016
Accepted After Revision: 05/09/2016
Aeluropus littoralis is a monocot halophyte grass and provide valuable genetic resources for understanding the molecular mechanisms of stress-responsive genes, and improving tolerance to abiotic stresses in economically important crops. In an attempt to identify salt stressed responsive genes, 154 isolated expressed sequence tags (EST) from A. littoralis were bioinformatically analyzed and functionally annotated. Of the 129 assembled unique transcripts, 111 (86%) and 18 (14%) comprised of singletons and contigs, respectively. Among them, 58.9% could be assigned a putative identity, 20.9% with hypothetical or unknown functions and 20.9% showed no match with existing sequences. Expression pattern of 41 selected ESTs were estimated by quantitative real-time polymerase chain reaction (qPCR) in two different tissues. Expression profiling were undertaken in control and three time point of salt stress (6hrs, 24hrs and one week) followed by three time point of recovery condition (6hrs, 24hrs and one week). In the root, the genes of SAMDC, ISB1 (6hrs), PP2C and SelO, HsfA1a, TFC D, Katanin, F-box were significantly up-regulated relative to control while LecRLKs, ARP, HP3, PICKLE, Utp20, SYP81, CIPK20, HAK18, VDAC3, SND1, NAP1, ISB1 (6hrs and 24hrs), NUC2, MUT, HP1 and PIP1;3 showed down-regulation in given conditions. In the case of leaf tissue, the genes of PP2C, SelO, Utp20, SND1, PITP, LecRLKs, STPK, KCNK12, HsfA1a, HAK18, NUC2, ARP, HP3 and ARP were significantly up- or down-regulated. Differential regulation of these genes were observed in root and tissue which confirm their role in salt stress tolerance. This functionally annotated EST and gene expression profiling provide initial insights into the transcriptome of A. littoralis.
Aeluropus Littoralis, Gene Expression, Salt Stress, Recovery Condition
Hashemipetroudi S. H, Nematzadeh G, Ahmadian G, Yamchi A, Kuhlmann M. Expression Analysis of Salt Stress Related Expressed Sequence Tags (Ests) from Aeluropus Littoralis by Quantitative Real-Time PCR. Biosc.Biotech.Res.Comm. 2016;9(3).
Hashemipetroudi S. H, Nematzadeh G, Ahmadian G, Yamchi A, Kuhlmann M. Expression Analysis of Salt Stress Related Expressed Sequence Tags (Ests) from Aeluropus Littoralis by Quantitative Real-Time PCR. Biosc.Biotech.Res.Comm. 2016;9(3). Available from: https://bit.ly/2BvESuD
Introduction
The global climatic changes, such as prolonged drought, temperature change and increasing salinity, cause to a serious challenge for agricultural production worldwide, affecting plant growth and yield. Drought and salinity are becoming particularly widespread in many regions, and may cause serious salinization of more than 50% of all arable lands by the year 2050. Therefore it is important to secure food production for a growing world population by increasing the yield of crop plants while resources become more restricted (Yamaguchi and Blumwald, 2005, Pitman and Läuchli, 2002 and Mittler and Blumwald, 2010).
The discovery of novel stress-responsive genes, identification of new cis-and trans- acting elements that are involved in stress adaptation provide an opportunity for generating stress tolerance crops (Cushman and Bohnert, 2000, Patnaik and Khurana, 2001, Ben-Saad et al., 2012 and Hashemi et al., 2016).
The use of wild plant species or halophytic relatives has been considered in plant breeding programs for developing salt and drought tolerant crops Utilising such approach, Aeluropus littoralis can serve as a halophyte model for identification and isolation of the novel adaptation genes. Aeluropus littoralis is a perennial monocot grass with the small haploid genome of 349 Mb, using the C4 mechanism for carbon fixation (Wang, 2004). Aeluropus littoralis grows in dry salty areas or marshes (Saad et al., 2011) and can survive where the water salinity is periodically high (Mesléard et al., 1993) and tolerate up to 1100 mM sodium chloride (Barhoumi et al., 2007). Therefore, A. littoralis serves as valuable genetic resource for understanding the molecular mechanisms of stress-responses in monocots, and can potentially be used for improving tolerance to abiotic stresses in economically important crops (Saad et al., 2010).
The process of identifying new genes and characterizing their functions generally is done at three molecular biology levels viz: genomics, transcriptomics and proteomics. Transcriptome-based gene discovery in response to environmental stress offers insights into the roles of the transcriptome in the regulation of physiological and biological responses (Gracey, 2007). Because these methods strictly clarify changes in transcript level, a complex multi-component process, such as salt and drought stress, can be broken into their basic element (Umezawa et al., 2002). Partial cDNA isolation often known as expressed sequence tags (ESTs) is the rapid and cost-effective Transcriptome-based gene discovery method that has become an efficient approach for identifying of coding regions in a wide spectrum of organisms.
Various techniques such as differential display PCR (DDPCR) (Hubank and Schatz, 1994), cDNA-amplified fragment length polymorphism (AFLP) (Bachem et al., 1996), suppression subtractive hybridization (SSH) (Diatchenko et al., 1996), serial analysis of gene expression (SAGE) (Velculescu et al., 1995), massively parallel signature sequencing (MPSS) and recently whole transcriptome profiling (RNA-Seq) (Brenner et al., 2000) have been used for EST isolation. By use of these techniques a large number of genes expressed during different developmental, differentiation and growth stages or in response to a variety of biotic and abiotic stresses has already identified in plants (Priya et al., 2012). It is clear now, the most biological processes, growth and developmental programming are regulated by the precise control of genetic expression (Agarwal et al., 2008). Genome-wide analyses of mRNA level showed that the expression level of genes may be changed (up or down-regulation) in response to different condition (Rabbani et al., 2003), in some case differential regulation of specific genes and pathways can lead to adaptation of crop genotypes to different abiotic stress (Aglawe et al., 2012).
To gain insight into these processes, it is necessary to study patterns of gene expression. Quantitative real-time polymerase chain reaction (qPCR) analysis is one of the most currently used approaches for measuring gene expression level (Gutierrez et al., 2008). The sensitivity, specificity and simplicity of this technique is incomparable with other methods such as Northern and in situ hybridization, RNase protection assays and semi-quantitative reverse transcription- polymerase chain reaction (RT-PCR) (Bustin, 2000).
In our pervious study (Fatemi et al 2016.), 154 ESTs (relative to salt and drought stresses) have been isolated from A. littoralis by cDNA-AFLP and their sequences were deposited in dbEST database (NCBI; www.ncbi.nlm.nih.gov/dbEST). The primary goal of this investigation was to annotate and assign putative functions of 154 isolated ESTs. Also, expression pattern of selected ESTs was analyzed in two different tissues of A. littoralis at salt stress and recovery condition.
Material And Methods
Aeluropus littoralis seeds were collected from Isfahan province (Roddasht region) in Iran and the sterilized seeds plated on full strength MS medium (Murashige and Skoog, 1962) with vitamins, 3% sucrose and 0.7% agar (pH 5.8). The cultures were incubated in germinator at 25 ± 2 C with 16 h light/8 h dark photoperiod at 100 μmol m-2 s-l photon flux density using cool-white fluorescent light. Two weeks after germination, the seedlings were transferred to hydroponic culture containing Hoagland’s solution (Hoagland and Arnon, 1950). The 30 day-old seedlings were stressed in 600 mM of sodium chloride at six passages (received 100 mM sodium chloride per 48 hrs up to 600 mM). At the end of the sixth passage, salt stress samples were collected at 6hrs (S1), 24hrs (S2) and one week (S3) time point. In order to plant recovery, the remained plants were transferred to a sodium chloride-free Hoagland’s solution, and then were collected after 6hrs (R1), 24hrs (R2) and one week (R3). Leaf and root were sampled in parallel. Control samples were taken from unstressed plants at the start of the experiment. All samples were immediately frozen in liquid nitrogen and stored at -70°C for RNA extraction.
The EST sequences of Aeluropus littoralis were retrieved from EST database at NCBI and were analyzed using the BLASTN, tBLASTX and BLASTX algorithms (Zhang et al., 2000). The database of gene ontology (http.//www.geneontology.org) was used to investigate the molecular function of each EST and its role in biological processes as well as its location in the cell. After selection of candidate reference genes, the gene-specific primers were designed using the Primer 3 software (Rozen and Skaletsky, 1999), and were synthesized by Metabion GmbH (Martinsried, Germany). All designed primers had 18-24 length, GC content ranging from 42% to 61% and similar melting temperatures (55-64°C). The amplicon length ranged from 60 to 282 bp. The primer sequences and GenBank accession numbers of related genes are presented in Table 1. The primer specificity was evaluated by melt curve analysis, and size of the amplicons was tested by end-point PCR on 3% agarose gels.
Total RNA was extracted using TRIzol reagent (Invitrogen Life Technologies, Karlsruhe, Germany) according to the manufacturer’s instructions. The quality and quantity of the extracted ribonucleic acid was checked by measuring absorbance at 260/280 nm using a NanoDrop spectrophotometer (Biochrom WPA Biowave II, UK). Further, the purity and integrity of RNA was tested by running on 1.2% agarose gel electrophoresis. Residual gDNA contaminating RNA extracts was removed by DNase treatment (DNase I RNase-free, Thermo Scientific, USA). The qPCR with three rDNA-based primers has recently been applied for DNA contamination assay by using RNA as template, (Hashemi et al., 2016).
The cDNA was synthesized using the QuantiTect reverse transcription kit (Qiagen) according to the manufacturer’s instructions. In brief, 1 μL (200 ng) of treated RNA, 1 μl of RT primer mix (blend of oligo-dT and random primers), 1 µl Quantiscript Reverse Transcriptase (contains RNase inhibitor), 4 μl Quantiscript RT Buffer 5X (includes Mg2+ and dNTPs) and 13 μl of RNase-free water were added and incubated at 65°C for 20 min and then followed by incubation at 95°C for 3 min for inactivation of reverse transcriptase. The final cDNA reactions were diluted 1:10, and stored at -20°C. Targets were amplified by the Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific) with two-step cycling in CFX96 real-time PCR instrument (Bio-Rad, USA) according to the company’s suggestions. The reaction master mix prepared by adding the following components: 1μL of cDNA (50 ng), 5 μl of 2X SYBR Green Master Mix and 0.3 μl of 10 μM of each primers and 3.4 μl of RNase-free water. Thermal cycling were performed using two-step cycling protocol according to the company’s procedures as follow: 10 min initial activation step at 95° C followed by 40 cycles of 95° C for 15 sec and 60° C for 1 min.
Data acquisition were performed during the annealing/extension step. After amplification, all PCR reactions were subjected to a thermal melt with continuous fluorescence measurement from 55°C to 95°C for dissociation curve analysis. Curves were analyzed by CFX Manager (Bio-Rad) with single threshold cycle and subtracted curve fit method. At least one non-template control (NTC) was used for each primer pair master mix. The threshold cycles (Ct) were automatically calculated for all reactions in the plate using the CFX manager software (Bio-Rad). All assays were carried out in three replications. The mean values for each assay were obtained, and used for further analysis. The livak (2 –∆∆Ct) method (Livak and Schmittgen, 2001) were used for calculation of relative gene expression ratio. RT2 Profiler PCR Array Data Analysis software (SABiosystems) was used to construct vocalno plot and clusterogram.
Results
Bioinformatic Analysis And Selecting Candidate Ests
In our pervious study, 154 A. littoralis ESTs from four different library were isolated by cDNA-AFLP method, and used in this study. EST sequences were retrieved through Entrez Gene -EST database- at the National Centre for Biotechnology Information (NCBI). The four libraries names were LIBEST_028119 (69 ESTs with GenBank accession number of JK191110.1-JK191042.1), LIBEST_027583 (25 ESTs with GenBank accession number of JK671243.1-JK671267.1), LIBEST_027584 (34 ESTs with GenBank accession number of JK671209.1-JK671242.1), and LIBEST_027576 (26 ESTs with GenBank accession number of JK671176.1-JK671201.1).
All ESTs were evaluated by the CAP3 DNA sequence assembly program. Assembling of the 154 ESTs produced a total of 18 assembled contigs and 110 singletons. Of the 129 assembled unique transcripts, 111 (86%) and 18 (14%) comprised of singletons and contigs, respectively. A total of 129 non-redundant dataset were compared to the GenBank non-redundant database using BLASTX to assign putative function. Of all aligned sequences, 58.9% could be assigned a putative identity, 20.9% with hypothetical or unknown functions and 20.9% showed no match with existing sequences. The size of most ESTs (79%) ranged from 100 to 400 bp. Sequence similarity searches using BLASTX were performed to compare the selected ESTs to subset of non-redundant protein sequences (nr) of green plant (taxid: 33090), Oryza sativa, Zea mays, Arabidopsis thaliana, Brachypodium distachyon, Setaria italica and Sorghum bicolor with several blast e-value cutoffs. We found that the A. littoralis transcript set (129 ESTs) showed a greater number of sequence similarity matches with Setaria italica and Zea mays transcripts. Based on functional annotation, 41 ESTs were selected for relative expression analysis in root and leaf tissues at different time-point of salt stress and recovery condition. The primer features are listed in Table 1.
| Table 1: showing the primer features | |||||
| Gene symbol | Accession number | Name | Function | E-value | Sequence |
| PIP3;1 | JZ191087 | Plasma membrane intrinsic protein | water channel activity | 3e-63 | TGTCATGGGCGTCTCCAAGT
GCAGTAGACGAGCGCGAAGA |
| VDAC3 | JZ191051 | Voltage dependent anion channel 3 | regulation of anion transmembrane transport | 1e-13 | TCCAGACCCAGCTGAAGCAC
GCCTGGTACACCAAGATCCTCA |
| SYP81 | JZ191048 | Syntaxin of plants 81 | Vesicle trafficking protein that functions in the secretory pathway. | 2e-32 | CAGCATGGCGTGGCTCTTAT
AGCATCTTGAAAGCGCATGG |
| NAP1 | JZ191072 | Nucleosome assembly protein1 | modulate chromatin structure by regulation of nucleosome assembly/disassembly | 0.29 | CAGGGCTCCACAAATCCAAC
ACGACCTGCTGAGTGCAAGC |
| CAND1 | JZ191057 | Cullin-associated and neddylation-dissociated | promotes the exchange of the substrate-recognition F-box subunit in SCF complexes | 2e-06 | TGGCAGTGACTACAGCATACGG
ACTGCGCACAGAGCGGTACT |
| SAMDC | JZ191058 | S-adenosylmethionine decarboxylase | Essential for polyamine homeostasis, and normal plant embryogenesis, growth and development. | 5e-14 | CCATCCATGGTCCTGCTTTC
GGGTTGAAGCCCATGACCTC |
| Katanin | JZ191064 | Katanin p80 WD40 | microtubule severing | 1e-61 | TGATCCCTCCCTTCCCAGTT
CCTGAGCGAATGCGTAAACC |
| F-box | JZ191080 | F-box protein | Unknown | 1e-60 | TGCCCATGAACCATTGTACG
GCCCTGCAGATCAGGTCAAC |
| SND1 | JZ191081 | Staphylococcal nuclease domain-containing protein 1-like | posttranscriptional gene silencing by RNA, response to salt stress | 1e-18 | GCGGATCTGGCAGTATGGAG
ACCGCTGCCTGAACAGACTT |
| GTF3C5 | JZ191082 | General transcription factor 3C polypeptide 5-like | Involved in RNA polymerase III-mediated transcription | 2e-37 | TTCCAAGTGGCCATCAGGTT
AAAGGGCTTCCTGCCTCTTG |
| ISB1 | JZ191092 | Importin subunit beta-1 | protein transporter activity | 1e-42 | GCTCCAGCCAAATGTCAAGC
GGTCTTGGTCAACAGCTTCAGG |
| NUC2 | JZ191093 | Nucleolin 2-like | Involved in pre-rRNA processing and ribosome assembly | 7e-06 | AAGTCCAGTGTTGCGGTTGC
CCGCATTTCTCTTCCCCTTC |
| GlyI | JZ191094 | Glyoxalase I | carbohydrate metabolic process | 0.079 | GTGGCATGGACTTGCTACGG
CCGTGGCATCACAGAGGATT |
| CIPK20 | JZ191099 | CBL-interacting protein kinase 20 | protein serine/threonine kinase activity | 2e-09 | CAGGAGATGAGGCCAGCACT
CTGTTGCTGTTGCTGCTTGG |
| HAK18 | JZ191100 | High-affinity potassium transporter | potassium ion transmembrane transporter activity | 7e-37 | GGCCAGACATTTCAGACCACA
AGCCCTGATGACCGTGTTTC |
| ZF30 | JZ191101 | Zinc finger CCCH domain-containing protein 30 | regulation of transcription | 3e-08 | GCTCTTGTTGGCTCCCCTCT
TCACCATTTACGCCCCAATC |
| URM12 | JZ191103 | Ubiquitin-related modifier 12 | involved in tRNA modification | 4e-17 | ACTGCGATTGGGAGCTGTGT
CGTGGAGATGAAGACCACCA |
| 5PTASE11 | Jk671224 | Inositol polyphosphate 5-phosphatase | response to abscisic acid, response to auxin, response to jasmonic acid | 6e-11 | CACATGGAACATGAATGGCAAG
TGAACTCCTTGCTCCGAAAAGA |
| PITP | Jk671260 | Sec14p-like phosphatidylinositol transfer family protein | transporter activity | 3e-55 | GAAAGTAAAGATTGCGGAGAC
GGGTGCGAACTCTGAAAC |
| SPIKE1 | Jk671264 | DOCK family guanine nucleotide exchange factor | vesicle-mediated transport | 1e-24 | TAAACAACACGGTGGCAGGTA
GCTCCCCATCAAATGTCCATA |
| TBC1 | Jk671226 | TBC1 domain family member 5 homolog B | act as a GTPase-activating protein for Rab family protein(s) | 3e-18 | CGGGATGGGAGCAACAAC
CACGGATAAGGGCACTGGT |
| TTL | Jk671266 | tubulin-tyrosine ligase | cellular protein modification process | 1e-05 | AAGAGGCAGTATCCTAATCAC
AAACTCATTCTGCCAATCTA |
| KCNK12 | Jk671259 | potassium channel subfamily K, member 12 | potassium ion transmembrane transport | 1.5 | TCGGAATCTGCCCTGAATCT
TATGTATCCCGGTCCACCACT |
| LecRLKs | JK671176 | G-type lectin S-receptor-like serine/threonine-protein kinase | Protein kinase activity | 5e-16 | CGGCCGACAATGGGTGAAG
GGGCATGCCAACCTCCTGTAG |
| SAP | JK671180 | Putative senescence-associated protein | – | 2e-11 | TGACACACCCCACACATACAA
GGTTTAGACCGTCGTGAGACAG |
| ARP | JK671182 | Auxin-repressed protein | – | 6e-08 | GGAAGTTTTGGGCTGTCTTTA
ATTTCGATGTTGCCTACTCTCTA |
| HP1 | JK671187 | Hypothetical protein1 | Similar to F-box family protein | 1.5 | CCAACAACTCAGCTCCAA
GATGTGAAAATAAGCACGCTA |
| MTL1 | JK671192 | Mitochondrial translation factor 1 | Mitochondrial protein translation and group II intron splicing | 6e-50 | ATTTCGGCAAAAGGAATGGAG
GAAGCTTGATGAGGCGACAGA |
| HP2 | JK671195 | Hypothetical protein2 | – | 0.006 | GTTTGGGCATTGGGTCCTCAAGT
CGAGCAACAGCAGCAAGAGCAC |
| MUT | JK671196 | Mutator-like transposase | Transposable element gene | 3e-18 | GATGCCCATCTTGACAATAC
GCAGTGGGGAAGTTGATTT |
| HP3 | JK671200 | Hypothetical protein3 | – | 0.003 | GGAAGTTTTGGGCTGTCTTTAC
AGATTTCGATGTTGCCTACTCTC |
| HsfA1a | Jk671211 | heat shock factor A1a | transcription factor activity | 4e-10 | GCAGTGCCCAGTTGTCTT
TTGGGCCTGGTGTCATA |
| PP2C | Jk671236 | Protein phosphatase 2C | protein serine/threonine phosphatase activity | 6e-34 | TAATATGCAGGGGAGGAAA
CAGCGAGTACACCACCAA |
| C-NAD-MDH2 | Jk671223 | Malate dehydrogenase | Cytosolic-NAD-dependent malate dehydrogenase 2 | 4e-20 | AAAACGTCGTTCAAAGAG
GCCATAAGATCCGTCAG |
| 5PTASE11 | Jk671224 | Inositol polyphosphate 5-phosphatase | response to abscisic acid, response to auxin, response to jasmonic acid | 6e-11 | CACATGGAACATGAATGGCAAG
TGAACTCCTTGCTCCGAAAAGA |
| HsfA1a | Jk671211 | heat shock factor A1a | transcription factor activity | 4e-10 | GCAGTGCCCAGTTGTCTT
TTGGGCCTGGTGTCATA |
| PP2C | Jk671236 | Protein phosphatase 2C | protein serine/threonine phosphatase activity | 6e-34 | TAATATGCAGGGGAGGAAA
CAGCGAGTACACCACCAA |
| C-NAD-MDH2 | Jk671223 | Malate dehydrogenase | Cytosolic-NAD-dependent malate dehydrogenase 2 | 4e-20 | AAAACGTCGTTCAAAGAG
GCCATAAGATCCGTCAG |
| PI/PC-TP | Jk671213 | Putative phosphatidylinositol/phosphatidylcholine transfer protein SFH8-like | phosphatidylinositol transporter activity, transporter activity | 6e-13 | TTGGCACATGCTTCCACATC
AGGACTGCCCCATCCATCAT |
| PICKLE | Jk671232 | CHD3-type chromatin-remodeling factor | DNA helicase activity | 1e-37 | AGGGGTATGCTGAACTTGT
CACCTTCGCCTCAATAA |
| RBPL39 | Jk671237 | RNA-binding protein 39-like | mRNA processing | 3e-05 | GGTGCCACTGGTCTGA
AAAGGGGAAGCTACAGGAG |
| SelO | Jk671243 | Selenoprotein O-like | transferase activity | 3e-13 | TCAAGGGTAGCGGAAAGAC
GGATGCTGCTGCGTAGAAC |
| TFC D | Jk671246 | tubulin folding cofactor D | GTPase activator activity | 4e-25 | TAAAAGATGCCGCAACATA GAAGGTGGGGAGCAAG |
| CHR11 | Jk671250 | chromatin-remodeling protein 11 | ATP-dependent chromatin remodeling, nucleosome binding | 7e-119 | CGCTGTTTTCTCTTTGATT
CGCTTTTGCCCTATTCTA |
| Utp20 | Jk671251 | small subunit processome component 20 homolog | rRNA processing, associates with U3 snoRNA. | 2e-60 | -CTTTCAGTTGCGTTTAGATGT
CGCTTTCAGAAGTGATAAGG |
| STPK | Jk671258 | Serine/threonine-protein kinase ULK4-like | protein kinase activity | 1e-32 | CATTTTCTGCCACTGTATCCT
ACTTTTACACAACCATGCTCC |
Dna Contamination Assay
For DNA contamination assay, the RNA samples have been tested by qPCR with. In our pervious study, a new procedure for testing DNA contamination was explained (Hashemi et al., 2016). To monitor residual gDNA contamination in RNA sample, the total RNA samples were examined by three rDNA-based primer pairs in qPCR. Generally, observation of any band on the agarose gel or melting curve peak in qPCR analysis were considered as gDNA contamination. In this study, all RNA samples were tested by this procedure, and DNase-free RNA was used for cDNA synthesis.
Primer Validation
The cDNA synthetized from control and treatment samples was also tested by qPCR. Pooled cDNA tissue samples containing equal amounts of the cDNA from control and treatment conditions were used to determine the primer pairs annealing temperature and their specificity. Primers annealing temperature were adjusted to 55- 60°C, and their specificity was checked by melt curve analysis and electrophoresis in 3% agarose gels. Single sharp peak with no primer-dimer was used for relative expression analysis. From 41 ESTs tested in the melt curve analysis, three of the genes (C-NAD-MDH2, TTL, SAP and PI/PC-TP) were not amplified and excluded from further analysis. The sharp peak of 5PTASE11 was only observed in root samples while no peak was detectable in leaf samples. The Ct value of CHR11 in root time point were higher than 35 and therefore excluded from analysis. For RBPL39 and HP2 genes, sharp peak were amplified only in leaf time points while unambiguous TFC D peak was only observed in root tissue. The trend of regulated genes in root and leaf tissues are presented in Figure 1.
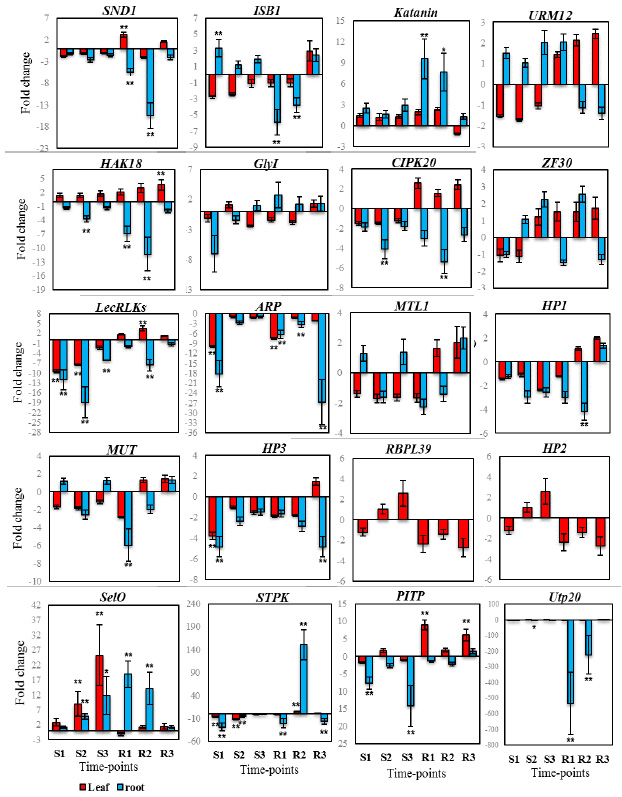 |
Figure 1: Trend of regulated genes during different time point of salt stress and recovery condition. |
Root Time Point Analysis
Distribution of Ct values amplified from root samples showed that in control and salt stressed samples, Ct value of most genes were lower than 25. Percent distribution of Ct values in Ct range of <25 in different time-points including control. Salt Stress: S1, S2, S3, Recovery condition: R1, R2 and R3 were 54.63%, 59.26%, 50.00%, 53.70%, 6.48%, 5.56% and 59.26%, respectively. These values indicated that the mRNA level of most genes in control and salt-stressed samples were higher than in the mRNA level of recovered samples (except R3). For normalization of expression levels in Aeluropus littoralis, different set of reference genes as well as their optimal number were recommended for root and leaf samples (Hashemi et al., 2016).
The three genes namely, RPS3, EF1A and UBQ were used as normalizer in root samples. For identification of genes with statistically significant gene expression changes, a volcano plot were used. Volcano plots are used to look at fold change and statistical significance, simultaneously (Allison et al., 2006).
In this study, expression values of 31 genes across six time point of root samples were compared to control samples by volcano plots (Figure 2). Values above the blue line and outside of the vertical lines were determined to statistically significant fold changes with 95% confidence (á=0.05). Genes with fold change higher than 2 or lower than -2 and p-value < 0.05 are indicated in blue in Figure 2.
Based on volcano plot analysis, seven genes including ZF30, URM12, CAND1, SPIKE1, TBC1, HP1, MTL1 and 5PTASE had not significant difference relative to control group. At time point S1, the expression level of SAMDC (3.1) and ISB1 (3.3) were higher while LecRLKs (-12.1), ARP (-18.2) and HP3 (-4.8) were downregulated (p-value < 0.05). Despite of the expression level of PP2C (4. 2) and GlyI (-7) were higher and lower than 2 and -2, but their fold changes were not significant (value inside parentheses is fold change). In S2 time point, two gene of PP2C (5.7) and SelO (4.8) were significantly upregulated while PICKLE (-4.5), Utp20 (-3.8), SYP81 (-3.6), CIPK20 (-4.1), HAK18 (-3.8) and LecRLKs (-18.9) were significantly downregulated (p-value < 0.05). PP2C (13.4) and SelO (11.9) and SAMDC (4.1) were upregulated in S3 time point while LecRLKs (-6.2) were significantly downregulated.
Under recovery conditions at time point R1, the expression level of HsfA1a (3.2), SelO (18.9), TFC D (81.1), SAMDC (11.1), Katanin (9.5), F-box (4.3) were increased relative to control while, the genes of Utp20 (-533.5), VDAC3 (-4.7), SND1 (-5.5), NAP1 (-10.5), ISB1 (-5.9), NUC2 (-11.7), HAK18 (-6.9), ARP (-6.3), MUT (-5.9) showed downregulation in mRNA level. The genes found to be upregulated at time point R2 were similar to that of time point R1. HsfA1a (3.4), SelO (14.1), TFC D (49.6), SAMDC (5.5), Katanin (7.6), F-box (3) showed upregulation in R2 time point. A number of eleven genes including Utp20 (-223.1), SYP81 (-5.7), SND1 (-15.4), NAP1 (-16.8), ISB1 (-3.7), NUC2 (-5), CIPK20 (-5.3), HAK18 (-11.3), LecRLKs (-7.6), ARP (-3.3), HP1 (-4.1) showed downregulation in R2 time point. In the R3 time point, three genes of PIP1;3 (-9.9), ARP (-26.8) and HP3 (-4.8) only showed downregulation in mRNA level.
Visualization of gene expression differences among different time point was done by RT2 Profiler PCR Array Data Analysis program (Figure 3 A). The result of the cluster analysis is depicted in a clustergram that shows all 31 genes analyzed and the magnitude each is expressed in control and 6 different root time points including group 1 (S1), group 2 (S2), group 3 (S3), group 4 (R1), group 5 (R2), group 6 (R3). Four major clusters of genes were observed: cluster I and II contained upregulated genes and cluster III and IV contained downregulated genes.
Leaf Time Points Analysis
Distribution of Ct values in leaf samples showed that the most of genes had Ct range of 25-30. Percent of Ct values in range of 25-30 in different time-points including control, S1, S2, S3, R1, R2 and R3 time-points were 57.41%, 61.11%, 42.59%, 48.15%, 50.00%, 45.37% and 54.63%, respectively. Comparing of Ct values in root and leaf samples showed that the mRNA level in root samples were higher than leaf samples. The geometric mean of U2SURP and GTF Ct values were chosen as normalizer in leaf samples.
Similar to root analysis, expression values of 31 genes across 6 time point of leaf samples were compared to control samples by volcano plots (Plots have not shown). Based on volcano plot analysis, expression level of 18 genes including ZF30, URM12, TBC1, HP1, MTL1, PICKLE, PIP1;3, VDAC3, SYP81, NAP1, CAND1, SAMDC, Katanin, F-box, ISB1, GlyI, CIPK20, HP2, MUT, RBPL39, SPIKE1 were unchanged relative to control group in different leaf time point.
In S1 time point, PP2C (3.4) were upregulated while NUC2 (-3.3), LecRLKs (-9.5), ARP (-9.9) and HP3 (-3.8) and STPK (-6.7) were significantly downregulated (p-value < 0.05). In S2 time point, SelO (8.9) were significantly upregulated while LecRLKs (-7.5), STPK (-12.3) and KCNK12 (-4.7) were significantly downregulated. In S3 time point, only upregulation of PP2C (3.4), SelO (8.9) and Utp20 (-533.5) were observed among all analyzed genes. PP2C (3.4), SND1 (3.1) and PITP (8.9) showed upregulation in R1 time point while ARP (-7.5) were significantly downregulated. In the R2 time point, the expression level of PP2C (6.5), LecRLKs (3.5), STPK (5.1) and KCNK12 (4.1) were significantly were increased relative to control. Finally, HsfA1a (3.4), HAK18 (-6.9), PITP (8.9) showed upregulation in R3 time point. The leaf clustergram were presented in Figure 3 B. Three major clusters of genes were observed. The most significantly up or down-regulated genes allocated into cluster II. This cluster represents genes reacting to recovery conditions.
 |
Figure 3: Clustergram of the magnitude of gene expression for 31 genes analyzed. Light green represents minimal gene expression, and red indicated maximum gene expression as indicated by the legend. |
 |
Figure 3b |
Discussion
The development of salt or drought-adopt crops either through the use of the crops wild relatives as genetic resources or domestication of naturally tolerant species have been proposed as a strategy to face with the environmental challenges. Halophytes as crops naturally salt-tolerant species are now being promoted in agriculture, particularly to provide forage, medicinal plants, aromatic plants (Flowers et al., 2010). Although, improving crop salt tolerance by genetic engineering is not easy, halophyte germplasm can furnish “climate-ready” genes for plant breeding program (Jaradat, 2010). Different aspects of A. littoralis properties such as life style, morphological, anatomical, ecological, physiological and molecular characteristics have been investigated so far (Hashemi et al., 2013, Hashemi-Petroudi et al., 2014).
In the present study, we have focused on gaining insight on differential regulation of some responsive ESTs in response to salt stress and recovery condition. Expression pattern of 41 selected ESTs were estimated by qPCR in root and leaf tissue. In the root, the genes of SAMDC, ISB1 (6hrs), PP2C and SelO, HsfA1a, TFC D, Katanin, F-box were significantly up-regulated relative to control while LecRLKs, ARP, HP3, PICKLE, Utp20, SYP81, CIPK20, HAK18, VDAC3, SND1, NAP1, ISB1 (6hrs and 24hrs), NUC2, MUT, HP1 and PIP1;3 showed down-regulation in given conditions. In the case of leaf tissue, the genes of PP2C, SelO, Utp20, SND1, PITP, LecRLKs, STPK, KCNK12, HsfA1a, HAK18, NUC2, ARP, HP3 and ARP were significantly up- or down-regulated. Interestingly, expression of some genes was induced by salt stress while also significantly repressed by recovery condition. Here interestingly the transcriptional regulator HsfA1a could be found. HsfA1a was formerly described to be a main component of the heat and drought stress response (Liu et al., 2013, Wang et al., 2015).
In this study, the protein phosphatase 2C (PP2C) were activated in salt stress and recovery condition. The PP2Cs from various organisms have been implicated to act as negative modulators of protein kinase pathways involved in diverse environmental stress responses and developmental processes (Xue et al., 2008). The SYP81 showed down regulation in given conditions. Syntaxins (with the exception of syntaxin 11) are transmembrane proteins which their functions respect to organism growth, physiology and development are not well known (Teng et al., 2001). The coexpression analysis showed that, the most significantly up or down-regulated genes allocated into cluster II (leaf tissue). Genes found in Cluster II are interesting candidates for physiological reactions related to the recovery of the plant after salt stress. This cluster represents genes reacting to recovery conditions.
Conclusion
In this study we bioinformatically analyzed the 154 ESTs from A. littoralis and the functionally annotation showed that 58.9% of ESTs had a putative function, 20.9% were hypothetical or unknown functions and 20.9% showed no match with existing sequences. The qPCR expression analysis of 41 selected ESTs showed different regulation in leaf and root tissue. The gene expression profiling has done in this study will also provide insight into the role of selected ESTs in different time points of salt stress and recovery condition. Differential regulation of these genes also point at their role in salt stress tolerance in plant. These information facilitate understanding the molecular mechanisms of stress related genes and could be used as valuable starting point for further research on these genes.
References
Agarwal, M., Shrivastava, N. & Padh, H. (2008). Advances In Molecular Marker Techniques And Their Applications In Plant Sciences. Plant Cell Reports, 27, 617-631.
Aglawe, S., Fakrudin, B., Patole, C., Bhairappanavar, S., Koti, R. & Krishnaraj, P. (2012). Quantitative Rt-Pcr Analysis Of 20 Transcription Factor Genes Of Mads, Arf, Hap2, Mbf And Hb Families In Moisture Stressed Shoot And Root Tissues Of Sorghum. Physiology And Molecular Biology Of Plants, 18, 287-300.
Allison, D. B., Cui, X., Page, G. P. & Sabripour, M. (2006.) Microarray Data Analysis: From Disarray To Consolidation And Consensus. Nature Reviews Genetics, 7, 55-65.
Bachem, C. W., Hoeven, R. S., Bruijn, S. M., Vreugdenhil, D., Zabeau, M. & Visser, R. G. (1996) Visualization Of Differential Gene Expression Using A Novel Method Of Rna Fingerprinting Based On Aflp: Analysis Of Gene Expression During Potato Tuber Development. The Plant Journal, 9, 745-753.
Barhoumi, Z., Djebali, W., Chaïbi, W., Abdelly, C. & Smaoui, A. (2007). Salt Impact On Photosynthesis And Leaf Ultrastructure Of Aeluropus littoralis. Journal Of Plant Research, 120, 529-537.
Ben-Saad, R., Ben-Ramdhan, W., Zouari, N., Azaza, J., Mieulet, D., Guiderdoni, E., Ellouz, R. & Hassairi, A. (2012). Marker-Free Transgenic Durum Wheat Cv. Karim Expressing The Alsap Gene Exhibits A High Level Of Tolerance To Salinity And Dehydration Stresses. Molecular Breeding, 30, 521-
533.
Brenner, S., Johnson, M., Bridgham, J., Golda, G., Lloyd, D. H., Johnson, D., Luo, S., Mccurdy, S., Foy, M. & Ewan, M. (2000). Gene Expression Analysis By Massively Parallel Signature Sequencing (Mpss) On Microbead Arrays. Nature Biotechnology, 18, 630-634.
Bustin, S. A. (2000). Absolute Quantification Of Mrna Using Real-Time Reverse Transcription Polymerase Chain Reaction Assays. Journal Of Molecular Endocrinology, 25, 169-193.
Cushman, J. C. & Bohnert, H. J. (2000) Genomic Approaches To Plant Stress Tolerance. Current Opinion In Plant Biology, 3, 117-124.
Diatchenko, L., Lau, Y., Campbell, A. P., Chenchik, A., Moqadam, F., Huang, B., Lukyanov, S., Lukyanov, K., Gurskaya, N. & Sverdlov, E. D. (1996). Suppression Subtractive Hybridization: A Method For Generating Differentially Regulated Or Tissue-Specific Cdna Probes And Libraries. Proceedings Of The National Academy Of Sciences, 93, 6025-6030.
Fatemi, F., Nematzadeh, G., Askari, H. & Hashemi, H.(2016) Differential Gene Expression Of Aeluropus Littoralis In Response To Nacl.Unpublished data.
Flowers, T. J., Galal, H. K. & Bromham, L. (2010). Evolution Of Halophytes: Multiple Origins Of Salt Tolerance In Land Plants. Functional Plant Biology, 37, 604-612.
Gracey, A. Y. 2007. Interpreting Physiological Responses To Environmental Change Through Gene Expression Profiling. Journal Of Experimental Biology, 210, 1584-1592.
Gutierrez, L., Mauriat, M., Pelloux, J., Bellini, C. & Van Wuytswinkel, O. (2008). Towards A Systematic Validation Of References In Real-Time Rt-Pcr. The Plant Cell Online, 20, 1734-1735.
Hashemi-Petroudi, S. H., Nematzadeh, G., Askari, H. & Ghahary, S. (2014). Involvement Of Cytosine Dna Methylation In Different Developmental Stages Of Aeluropus littoralis. Journal Of Plant Molecular Breeding, 2, 56-67.
Hashemi, S. H., Nematzadeh, G., Ahmadian, G., Yamchi, A. & Kuhlmann, M. (2016). Identification And Validation Of Aeluropus littoralis Reference Genes For Quantitative Real-Time Pcr Normalization. Journal Of Biological Research-Thessaloniki, 23, 1.
Hashemi, S. H., Nematzadeh, G., Askari, H. & Ghasemi, Y. (2013). Pattern Of Dna Cytosine Methylation In Aeluropus littoralis During Temperature Stress. Journal Of Plant Molecular Breeding, 1, 16-24.
Hoagland, D. R. & Arnon, D. I. (1950. The Water-Culture Method For Growing Plants Without Soil. Circular. California Agricult)ural Experiment Station, 347.
Hubank, M. & Schatz, D. (1994) Identifying Differences In Mrna Expression By Representational Difference Analysis Of Cdna. Nucleic Acids Research, 22, 5640-5648.
Jaradat, A. A. (2010). Genetic Resources Of Energy Crops: Biological Systems To Combat Climate Change. Australian Journal Of Crop Science, 4, 309.
Liu, Y., Zhang, C., Chen, J., Guo, L., Li, X., Li, W., Yu, Z., Deng, J., Zhang, P., Zhang, K. & Zhang, L. (2013). Arabidopsis Heat Shock Factor Hsfa1a Directly Senses Heat Stress, Ph Changes, And Hydrogen Peroxide Via The Engagement Of Redox State. Plant Physiol Biochem, 64, 92-8.
Livak, K. J. & Schmittgen, T. D. (2001). Analysis Of Relative Gene Expression Data Using Real-Time Quantitative Pcr And The 2− Δäct Method. Methods, 25, 402-408.
Mesléard, F., Ham, L. T., Boy, V., Van Wijck, C. & Grillas, P. (1993). Competition Between An Introduced And An Indigenous Species: The Case Of Paspalum paspalodes (Michx) Schribner And Aeluropus ittoralis (Gouan) In The Camargue (Southern France). Oecologia, 94, 204-209.
Mittler, R. & Blumwald, E. (2010). Genetic Engineering For Modern Agriculture: Challenges And Perspectives. Annual Review Of Plant Biology, 61, 443-462.
Murashige, T. & Skoog, F. (1962). A Revised Medium For Rapid Growth And Bio Assays With Tobacco Tissue Cultures. Physiologia Plantarum, 15, 473-497.
Patnaik, D. & Khurana, P. (2001). Wheat Biotechnology: A Minireview. Electronic Journal Of Biotechnology, 4, 7-8.
Pitman, M. G. & Läuchli, A. (2002). Global Impact Of Salinity And Agricultural Ecosystems. Salinity: Environment-Plants-Molecules. Springer.
Priya, A., Tripathi, H., Yadav, D. K., Khan, F., Gupta, V., Shukla, R. K. & Darokar, M. (2012). Functional Annotation Of Expressed Sequence Tags Of Papaver somniferum. Plant Omics, 5, 223.
Rabbani, M. A., Maruyama, K., Abe, H., Khan, M. A., Katsura, K., Ito, Y., Yoshiwara, K., Seki, M., Shinozaki, K. & Yamaguchi-Shinozaki, K. (2003). Monitoring Expression Profiles Of Rice Genes Under Cold, Drought, And High-Salinity Stresses And Abscisic Acid Application Using Cdna Microarray And Rna Gel-Blot Analyses. Plant Physiology, 133, 1755-1767.
Rozen, S. & Skaletsky, H. (1999). Primer3 On The Www For General Users And For Biologist Programmers. Bioinformatics Methods And Protocols. Springer.
Saad, R. B., Romdhan, W. B., Zouari, N., Azaza, J., Mieulet, D., Verdeil, J.-L., Guiderdoni, E. & Hassairi, A. (2011). Promoter Of The Alsap Gene From The Halophyte Grass Aeluropus littoralis Directs Developmental-Regulated, Stress-Inducible, And Organ-Specific Gene Expression In Transgenic Tobacco. Transgenic Research, 20, 1003-1018.
Saad, R. B., Zouari, N., Ramdhan, W. B., Azaza, J., Meynard, D., Guiderdoni, E. & Hassairi, A. (2010) Improved Drought And Salt Stress Tolerance In Transgenic Tobacco Overexpressing A Novel A20/An1 Zinc-Finger “Alsap” Gene Isolated From The Halophyte Grass Aeluropus littoralis. Plant Molecular Biology, 72, 171-190.
Teng, F. Y. H., Wang, Y. & Tang, B. L. (2001). The Syntaxins. Genome Biology, 2, 1.
Umezawa, T., Mizuno, K. & Fujimura, T. (2002). Discrimination Of Genes Expressed In Response To The Ionic Or Osmotic Effect Of Salt Stress In Soybean With Cdna‐Aflp. Plant, Cell & Environment, 25, 1617-1625.
Velculescu, V. E., Zhang, L., Vogelstein, B. & Kinzler, K. W. (1995). Serial Analysis Of Gene Expression. Science, 270, 484-487.
Wang, R. (2004). Plant Functional Types And Their Ecological Responses To Salinization In Saline Grasslands, Northeastern China. Photosynthetica, 42, 511-519.
Wang, Y., Cai, S., Yin, L., Shi, K., Xia, X., Zhou, Y., Yu, J. & Zhou, J. (2015) Tomato Hsfa1a Plays A Critical Role In Plant Drought Tolerance By Activating Atg Genes And Inducing Autophagy. Autophagy, 11, 2033-2047.
Xue, T., Wang, D., Zhang, S., Ehlting, J., Ni, F., Jakab, S., Zheng, C. & Zhong, Y. (2008). Genome-Wide And Expression Analysis Of Protein Phosphatase 2c In Rice and Arabidopsis. Bmc Genomics, 9, 1.
Yamaguchi, T. & Blumwald, E. (2005). Developing Salt-Tolerant Crop Plants: Challenges And Opportunities. Trends In Plant Science, 10, 615-620.
Zhang, Z., Schwartz, S., Wagner, L. & Miller, W. (2000) A Greedy Algorithm For Aligning Dna Sequences. Journal Of Computational Biology, 7, 203-214.

