Department of Chemical Engineering, Beant College of Engineering & Technology, Gurdaspur 143521, Punjab, India
Article Publishing History
Received: 13/02/2016
Accepted After Revision: 23/03/2016
Sequence alignment is a prerequisite for biological sequence data analysis. In this paper, a systematic approach used to analyze the four proteins from Viral Capsid of Human Herpes Virus (HSV) which cause cytomegalovirus, brain inflammation, and lifelong infection. It is not viable to extend the relationship between drug-viral protein interactions of individual protein of HSV. With objective to develop the broad spectrum antibiotics, it is necessary to analyze the protein alignment and similarity. So the various point accepted scoring matrices (PAM) used for Capsid protein sequence alignment using Bioinformatics Tool of Matrix laboratory (Mat lab). This paper highlights the optimization of scoring matrices of aligned sequences and best scores for the different strain of HSV. From results showed that lower PAM matrix is suitable for capsid proteins of HSV-1 and HSV-2 due to closely related proteins of HSV. The results of sequence alignment can assist in the drug development and help to reduce the infection using antiviral therapy.
Human Herpes Virus, Sequence Alignment, Mat Lab And Scoring Matrices
Sohpal V. K. Computational Approach for Sequence Alignment of Capsid Proteins of Human Herpes Virus. Biosc.Biotech.Res.Comm. 2016;9(1).
Sohpal V. K. Computational Approach for Sequence Alignment of Capsid Proteins of Human Herpes Virus. Biosc.Biotech.Res.Comm. 2016;9(1). Available from: https://bit.ly/32pPGWU
Introduction
A wealth of molecular data concerning the function and structure of proteins and nucleic acids is available in the form of DNA, RNA, and protein sequences. Score from the sequence alignment has become an essential and widely used tool for understanding the functioning and phylogenetically divergent of different strains. Various scoring matrices (PAM, BLOSUM, and Gonnet) applications are used with Smith-Waterman and Needleman-Wunsch algorithm for sequence alignment. Point Accepted Mutations (PAM) matrix has significant advantages over the Blosum and Gonnet. PAM matrix is calculated by observing the differences in closely related proteins. In this paper, I have focused on HSV-1 and 2, which are two species of the herpes family, which cause infections in humans. Five proteins are from the Viral Capsid UL6, UL18, UL35, UL38 and Major Capsid proteins UL19 are from long unique region of HSV genome. It is most significant to study the sequence alignment and drug design. For this purpose optimization of scoring, matrices are used. The data extract using Bioinformatics toolbox of Matrix Laboratory have been used for a functional task of providing annotation of biologically relevant information from a nucleotide or proteomic sequence. It provides a powerful interface for the analysis and mining of genomic information while seamlessly handling the nonscientific complexities of interfacing with hardware, computational clusters, software packages, raw data, and file formats. Genome and proteome analysis performed using bioinformatics toolbox that extends MATLAB to provide an integrated software environment. An open prototype idea and extendable environment for MATLAB using efficient processing and statistical functions, a template for improving or creating and analyzing biological data.
In literature review our main focus was on the three principles that are necessary for studying and investigating the sequence alignment and phylogenetic analysis of human herpes virus. Firstly bioinformatics analyses, sequence alignment and phylogenetic analysis from biological databases sources have been studied. Secondly approaches, particularly with reference to herpes Virus have been attempted. At last the systematic analysis of the bioinformatics tools and softwares have also commonly been used for sequence alignment.
Biopipe framework that allows the researchers to focus on their specific biological analysis and avoid to deals with issues like data access and parsing and job management (Shawn, 2003). A new program has been developed for creating multiple alignments of protein sequences, of the algorithm and showing MUSCLE to achieve the highest scores reported to date on four alignment accuracy benchmarks (Edgar, 2004). It integrates the sources for genome annotation, inference of molecular interactions across species, and gene-disease associations in form of Atlas. It is based on relational data models that developed for each of the source data types. They have developed a new web application MIGenAS for processing basic bioinformatics tasks as well as orchestrating them into complex workflows within a single, coherent web interface Biowep, a web based client application that allows for the selection and execution of a set of predefined workflows accessed. It includes a workflow manager, a user interface and a workflow executor, (Rampp, 2006, Shaw, 2005 and Romano, 2007).
The Molecular Evolutionary Genetics Analysis software is a desktop application designed for comparative analysis of homologous gene sequences either from multigene families or from different species with a special emphasis on inferring evolutionary relationships and patterns of DNA and protein evolution. MEGA provides many convenient facilities for the assembly of sequence data sets from files or web-based repositories. It includes tools for visual presentation of the results obtained in the form of interactive phylogenetic trees and evolutionary distance matrices, (Kumar, et al., 2004; 2008).
Statistical score has been used for assessing the quality of multiple sequence alignments, where the quality assessment is based on counting the number of significantly conserved positions in the alignment using importance sampling method in conjunction with statistical profile analysis framework (Virpi, 2006). Aligning Sequences by minimum description length in which alignment algorithm uses minimum description length to encode and explore alternative expressions. The expression with the shortest encoding provides the best overall alignment (Conery, 2007).
These authors have worked on dynamic use of multiple parameter sets in sequence alignment. They have used an alignment algorithm to allow dynamic use of multiple parameter sets with different levels of stringency in computation of an optimal alignment of two sequences. Various workers have also developed techniques to assess the scores using splitting the BLOSUM score into numbers of biological significance. Kalign is an accurate and fast multiple sequence alignment algorithm. They developed Kalign, a method employing the Wu-Manber string-matching algorithm, to improve both the accuracy and speed of multiple sequence alignment, (Lassmann, 2005, Xiaoqiu, 2007 and Francesco,2007).
MAFFT has been used for improvement in accuracy of multiple sequence alignment. These new options of MAFFT showed higher accuracy than currently available methods including TCoffee version 2 and CLUSTAL W in benchmark tests consisting of alignments of more than 50 sequences.Herpes simplex virus type 2 UL56 interacts with the ubiquitin ligase and increases ubiquitination, (Katoh, 2005 and Ushijima, 2008).
The above work was concentrated on HSV-2 UL56 protein (UL56). Herpes simplex virus type 1 infection associated with atrial myxoma has been studied by Li, (2003). The binding partners for the UL11 tegument protein of herpes simplex virus type 1. They also worked on product of the UL11 gene of herpes simplex virus type-1 (Loomis, 2008). Worked and significance on protein interaction to functional annotation in herpes virus using graph alignment (Kolar, 2008). Simulate the function analysis of protein for distance of proteins using various tools (Vipan, 2012). READemption pipeline takes care of individual tasks and integrates them into an easy-to-use tool with a command line interface. It was mainly developed for the analysis of bacterial primary transcriptomes (Förstner, 2014). PiPipes used to analyze piRNA and transposon-derived RNAs from a variety of high throughput sequencing libraries, including small RNA, RNA, degradome or 7-methyl guanosine cap analysis of gene expression (CAGE), chromatin immuno-precipitation and genomic DNA-seq (Han et al.,2015).
From previously literature on optimization of scoring matrices using bioinformatics tools, reveals that a significant work has not been published. This paper is an attempt to blend wet biological lab data analysis and utilization of recently developed software for sequence alignment. The accuracy of sequence alignment is function of comparison of particular sequences for local and global alignment scores.
Material And Methods
Firstly the Protein sequence data of HSV-1 and HSV-2 had been accessed from the National Centre for Biotechnology Information (NCBI) and protein database bank of uniport KB. he retrieval of data on the basis of human herpes strain and respective accession number using matrix laboratory. The data from database bank for five proteins of the viral Capsid UL6, UL18, UL35, UL38 and major Capsid protein UL19 used for sequence alignment through MEGA 4. Secondly the programme had been developed in (Bioinformatics Tool Box) to compare the sequences of HSV for various point accepted mutation matrices using Needleman-Wunsch (NW) algorithm. The programmes compare the sequence and determine the optimal scoring matrices with score bits.
Results And Discussion
The developed algorithmic technique and statistic of sequence alignment helped to make optimal alignment and act as a valuable tool in bioinformatics for valid alignment. Optimal alignment serves to judge the similarity of sequence aligned. The statistical assessment of optimal alignment score makes sequence alignment less dependent on gap penalty choice. The alignment of HSV strain performed on different scoring matrix of PAM using Needleman-Wunsch algorithm. An algorithm, with MEGA and Bioinformatics tool box had been used to compare the point accepted mutation matrices and optimize it for four viral Capsid protein of HSV and one major Capsid protein. Before sequence alignment of protein Sequence Identity (number of exact matches divided by total sequence length) and Similarity (partial score given for similar amino acids of both strain HSV. The sequence identity of HSV-1 and HSV-2 strain (UL-19) for Major Capsid protein is (1292/1374) 94% and similarity of sequence 99%.Compartive sequence alignment indicates that highest similarity towards phylogenetic evolution of HSV-1 [NP_044620.1 and UniProt/Swiss-Prot P06491] and HSV-2 [NP_044488.1 and UniProtKB/TrEMBL P89442 ] strain using Needleman Wunsch
algorithm.
The 1769 protein of both strain (HSV-1 & 2) aligned for global alignment score with gaps. The alignment score is the function of aligned pairs with gap penalty and gap extension. The higher the alignment score, the better the alignment accuracy. Figure 1 indicates that alignment score for various PAM scoring matrices of Major capsid protein (UL-19) is decreased from left to right. It suggests that lower scoring matrices most suitable for this particular protein. As preceded from lower to higher value of PAM matrices, score bits reduced. The optimized score for major capsid protein is 5.0995e3 at 10 PAM value. The 1769 protein of HSV-1 aligned with 1772 protein of other two strain of virus. On aligning the other strains of HSV with HSV-1, the score bits are remaining same. This indicates that the HSV-3 and HSV-4 both are identical and play the same role in DNA processing and packaging. Although both have relatively same score but lower than HSV-1 and 2.The optimized score for another strain is 4.374 e3 and 4.375 e3 at 10 PAM value. The highest score (bits) corresponding PAM value is recommendable for major capsid protein. From the result and comparison, it is clear that HSV-3 and HSV-4 have same protein structures, which cause the same type of infection.
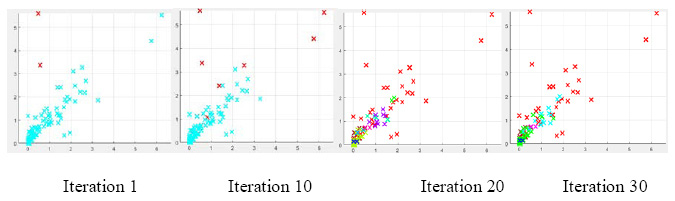 |
Figure 1: Major Capsid Protein of HSV-1 strain with other strain of HSV |
The Capsid protein of HSV virus is controlled by UL-18 gene, which play a role in replication of genetic material and facilitates to interact with glycoprotein. The global alignment score is reduced to one-third of Major Capsid protein of HSV-1 with HSV-2. According to the biostatics higher alignment score, gives the better the alignment. Figure 2 replicates that alignment and similarity of Capsid protein are at low. The global alignment score also decreases with PAM matrix. It means the lowest PAM matrices are suitable for alignment of Capsid protein. The optimum score for alignment of HSV-1 [P10202] and HSV-2 [P89441] strain is 1.1235 e 3 at 10 PAM. UL-38 genes is responsible for Small Capsid protein generation in HSV virus. The small Capsid protein has main function in Capsid assembly and the DNA maturation. Capsid assembly is the primary cause of viral infection through host cell. The DNA maturation takes place in an infected cell of the host. Small Capsid protein of HSV-1 [NP 044637] and HSV-2 [NP 044505] strains have lower score. It signified that protein has poor alignment and global alignment score is also low. Similarly the HSV-1 and HSV-3 (NP 040146) alignment is insignificant because the maximum score 45 at higher PAM matrices. The optimum score for an alignment of HSV-1 and HSV-2 strain is 3.523e2 with 10 PAM. On another hand, optimum score of HSV-1 and HSV-3 strain is 4.532e1 at matrix 350 and is shown in figure 3.
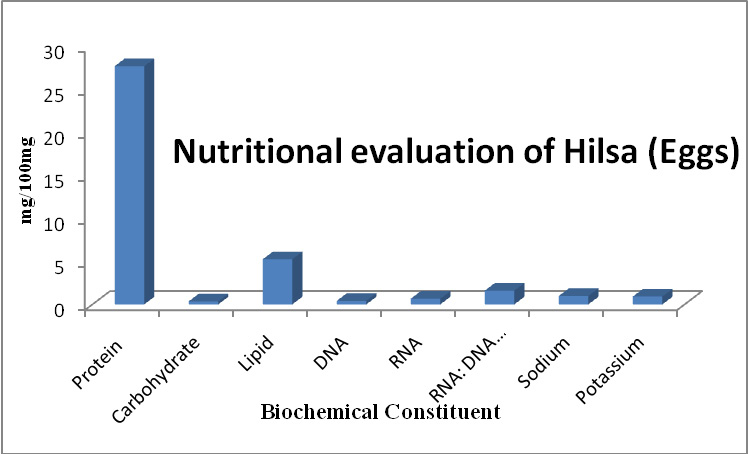 |
Figure 2: Capsid Protein of HSV-1 with HSV-2 strain |
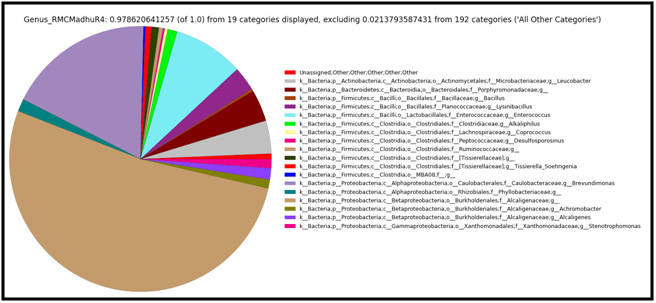 |
Figure 3: Small Capsid Protein of HSV-1 strain with other strain of HSV |
UL-6 associates with a UL-15/UL-28 protein complex during capsid assembly. The UL-15/UL-28 is believed to bind with viral DNA and serve the same purpose as terminate by packing viral DNA into the capsid during capsid assembly. The sequence alignment between HSV-1 and HSV 2 is higher at lower PAM value. While the global alignment score for HSV-3 strain is started with lower score and increases 100 PAM matrices. Contrary of earlier strain, HSV-5 has highest value higher PAM matrices. From figure 4, it is concluded that protein sequence similarity of HSV-1 and HSV-2 is highest. The optimum score for Capsid Portal Protein is 2.135e3, lowest for HSV-5 3.253e2 and intermediate for HSV-3 strain.
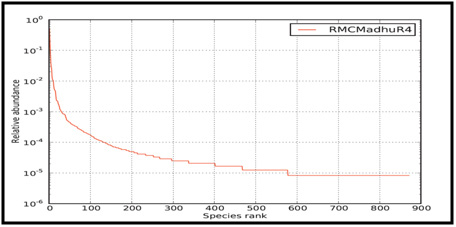 |
Figure 4: Capsid Portal Protein of HSV-1 strain with other strain of HSV |
Conclusion
Global alignment score versus PAM matrices profile can use to analysis the optimum score at particular PAM matrices. The optimized scores for major capsid protein, capsid protein and small capsid protein are 4.374 e3, 1.1235 e3 3.523e2 respectively at PAM value of 10. From the result, it found that lower PAM matrix is suitable for HSV-1 and HSV-2, while for other strains its value below is 200 PAM for higher global alignment score. From the results, it concludes that lower PAM matrices are highly suitable for conservative regions of sequence and vice versa.
References
Ahola, V Aittokallio T., Vihinen M. and Uusipaikka E. (2006). A statistical score for assessing the quality of multiple sequence alignments. BMC bioinformatics 7(1): 484.
Conery J.S. (2007). Aligning sequences by minimum description length. EURASIP Journal on Bioinformatics and Systems Biology, 4
Edgar R.C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity, BMC Bioinformatics 35(5): 1792-1797
Fabris F., Sgarro A. and A. Tossi (2007). Splitting the BLOSUM score into numbers of biological significance. EURASIP Journal on Bioinformatics and Systems Biology.
Förstner K.U., Vogel J. and Sharma C.M. (2014). READemption–A tool for the computational analysis of deep-sequencing-based transcriptomes data. Bioinformatics btu533.
Han, Bo W., W.Wang PD Zamore and Z.Weng (2015) piPipes: a set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome-and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics 31.4 : 593-595.
Huang X and Brutlag D.L. (2007). Dynamic use of multiple parameters sets in sequence alignment, Nucleic Acids Res. 35(2): 678–686.
Katoh K., Kuma K., Toh H. and Miyata T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment, Nucleic Acids Research 33: 511–518.
Kolá M., Lässig M. and Berg J. (2008). From protein interactions to functional annotation: graph alignment in Herpes. BMC systems biology 2(1): 90.
Kumar S., Nei M., Dudley J. and Tamura K. (2008). MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinformatics 9(4): 299–306.
Kumar S., Tamura K., Nei M. (2004). MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Briefing Bioinformatics 5(2): 150-163.
Lassmann T. and Sonnhammer E.L.L. (2005). Kalign – an accurate and fast multiple sequence alignment algorithm ,BMC Bioinformatics 6(2): 2105-6-298.
Li Y., Pan Z., Ji Y., Sheppard M., Jeffries D.J., Archard L.C. and Zhang H. (2003). Herpes Simplex Virus Type 1 Infection Associated with Atrial Myxoma, American Journal of Pathology 163(6): 2407–2412.
Loomis. Joshua S., Richard J. Courtney, and John W. Wills (2008). Binding Partners for the UL11 Tegument Protein of Herpes Simplex Virus Type 1. Journal of Virology 77(21): 11417–11424.
Rampp M., Soddemann T. and Lederer H. (2006). The MIGenAS integrated bioinformatics toolkit for web-based sequence analysis, Nucleic Acids Research 34(1): W15–W19
Romano P., Bartocci E., Bertolini G., De Paoli F., Marra D., Mauri G., Merelli E. and Milanesi L. (2007). Biowep: a workflow enactment portal for bioinformatics applications, BMC Bioinformatics 8(S19): 2105-2108
Shaw S.P., Huang, Y., Xu T., Yuen M. M., Ling J. and Ouellette B. F. (2005). Atlas–a data warehouse for integrative bioinformatics. BMC bioinformatics 6(1): 34.
Shawn H., Ratnapu K.K., Jer-Ming C., Kumarasamy B., Juguang X., Clamp M., Stabenau A., Potter S., Clarke L., Stupka E. (2003) Biopipe: A Flexible Framework for Protocol-Based Bioinformatics Analysis, Genome Res 13: 1904–1915
Ushijima Y., Koshizuka T., Goshima F., Kimura H. and Nishiyama Y. (2008). Herpes Simplex Virus Type 2 UL56 Interacts with the Ubiquitin Ligase Nedd4 and Increases Its Ubiquitination, Journal of Virology 82(11): 5220–5233.
Vipan.K S., Apurba D., Amarpal S., (2012). Comparative Analysis of Non-Synonymous and Synonymous Substitution of Capsid Proteins of Human Herpes Virus. J Proteomics Bioinformatics 5(8): 172-176.

