METS Institute of Pharmacy, Bhujbal Knowledge City, Adgaon, Nashik, Maharashtra, India.
Correspondig author email: jivanpatil4512@gmail.com
Article Publishing History
Received: 25/06/2022
Accepted After Revision: 14/08/2022
The rationale behind writing this review article is to give an introduction to the clinical trial, its phases and the current scenario in clinical research in India. This article gives a brief idea about the clinical trial phases. The reader can find the process of trial in step-by-step manner with the government regulatory body that has a major role to ensure the safety of the subject involved in the experimental study, with appropriate protocol and approval of the whole experimental study. This paper describes the role of the ethics committee, the investigator and sponsors’ responsibilities along with DCGI workflow, regulations of study. This study also discusses what is CDSCO, ICH-GCP government body in clinical trials etc. The clinical trial has a crucial role in serving good health to the public and developing new promising drug candidates in the treatment of diseases.
The new drug candidate and therapy enhance the quality and lifespan of the patient. Nowadays the number of clinical trials has increased in biomedical research so there is a huge need to make transparency and easy accessibility of trial studies to general people. Hence, the development of CTRI has been done. The regulatory authority strictly observes whether the guidelines are properly followed in trial or not. The regulatory guidelines are modified timely. The serious injury during trial and informed consent form are recently modified. This review article put all the essential things for the reader to get enough idea about a clinical trial in India, how it is conducted, which regulatory body involved in clinical trial etc.
Clinical Trial, CTRI, DCGI, Ethics Committee, ICH-GCP.
Pathak D. D. R, Sonawane S. S, Patil J. G, Kasar D. S, Chavan S. R, Shahane N. R, Gadak P. M, Shinde P. R, Kandalkar T. D. Clinical Trial Phases and their Registration in India: A Systematic Review. Biosc.Biotech.Res.Comm. 2022;15(3).
Pathak D. D. R, Sonawane S. S, Patil J. G, Kasar D. S, Chavan S. R, Shahane N. R, Gadak P. M, Shinde P. R, Kandalkar T. D. Clinical Trial Phases and their Registration in India: A Systematic Review. Biosc.Biotech.Res.Comm. 2022;15(3). Available from: <a href=”https://bit.ly/2U8EBeg”>https://bit.ly/2U8EBeg</a>
Copyright © This is an Open Access Article distributed under the Terms of the Creative Commons Attribution License (CC-BY). https://creativecommons.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provided the original author and sources are credited.
INTRODUCTION
This review gives an overview of clinical trials and the phases involved in a clinical trial. The reader can understand the clinical trial with its importance in India. The regulation of trials in India and the various government bodies that play important roles in the trial are described in this paper. Talking about pre-clinical research gave a satisfactory statement about drug safety but it did not give any clue regarding drug interaction with the human system or its overall action on humans. Clinical research offers the study of the drug on the human body, for this, researchers develop the clinical study design with various aspects to perform. There are clinical research phases and can initiate the investigational new drug process. This is the step that must be done before commencing clinical research.
The design of clinical research is made in such a way that it answers the questions related to drug products. For this, it is necessary to implement the plan and this plan is called PROTOCOL. The protocol can be made by either researchers or the manufacturer of the drug. For starting this clinical trial researchers must have basic pre-information regarding the drug and how the study be conducted to get a satisfactory result. Designing of the clinical study is nothing but, Researches have to decide basic things like duration of study, selection criteria for the subject i.e., who can involve in the study, the participants, the control group to avoid bias in future and most importantly drug administration to the subject, lastly the evaluation of data obtained. The clinical study was performed in four phases (Mahan 2014; FDA 2020).
A clinical trial is a scientifically controlled study which assess drug safety and efficacy in the human subject. This study has many pros, developing a new treatment that has a big beneficial effect over already existing therapy, the new treatment is equal to standard treatment etc (Mahan 2014). The role of the US-FDA begins after the completion of pre-clinical evaluation and safety. This practice must be stuck to good clinical practices (GCP) (Browne et al. 2014). The criteria that must be assured during the experimental study include the well-being of the human subject, sampling, range of masking, randomization trial i.e., RCT, assessing endpoints, and evaluation of results. All of these are necessary parameters to assess the effectiveness of drugs and also provide information related to adverse drug effects (Ambroz et al. 1981; Haahr et al. 2006; Esource.com 2022).
The RCT study involves study design like the parallel, cross-over, cluster or factorial study design. Likewise, evaluation of the hypothesis involves superiority and non-inferiority (Esource.com 2022). In this study, people were randomly assigned to different treatments under the experiment. The ideal randomization increases the statistical result and reduces the bias (Arvins 1998). A clinical trial involves different phases that are- phase I, phase II, phase III and phase IV. Phase I is also known as the “earliest phase” phase II also known as the exploratory trial, phase III is known as the “later phase” and phase IV is called the post-marketing phase. There is one more phase that happens at an earlier stage of all known as phase 0. In this phase drug, ADME and pharmacodynamic studies are performed (Esource.com 2022).
In phase I, there is the assessment of drug safety, in phase II effectiveness of the drug, and finally, the affirmation of both safety and effectiveness are assessed in phase III. Phase IV is the post-marketing survey that is also known as Pharmacovigilance (DeMets et al. 2015; Esource.com 2022). There is a necessity for conducting a clinical trial, to ensure the drug safety, quality and betterment of the patient’s life. There is an elevation in conducting a clinical trial in India. The primary duty of the government is to ensure the well-being of the human subject that participate in the trial experiment and to maintain international standards. The clinical trial conducted in India should be as per ICH guidelines and follow schedule Y.
Table 1. Showing the overview of clinical trial phases:
| Clinical trial phase | Description | Reference |
| Phase 0 | This phase is also known as the exploratory trial. This phase lack therapeutic and diagnostic goal. Phase 0 involves less no. of human exposure i.e., about 10 human subject.
Duration – 1 week Human participants – 10 Drug dose- sub therapeutic Study – pharmacokinetic & pharmacodynamic This study commence before the study of dose elevation, safety criteria and tolerance. Advantages: This study useful to remove drug therapy before going to phase second, it is used to evaluate new drug ADME and pharmacodynamics in humans, help to minimize the drug development expenses and time Disadvantages: This phase does not have any potential therapeutic goal, lack of active involvement of human subject to participate; it may delay sometimes or exclude patients from other clinical trials that may possess potential of therapeutic goal. Un-availability of sensitive analytical method this may cause limitation to this trial, Because it necessary in micro dosing. |
(Collin et al. 2007; Marchetti et al. 2007; Le Tourneau et al. 2009). |
| Clinical trial phase | Description | Reference | |||||
| Phase I | This phase is used to assess the administration of drug dose with repetition criteria, it also used assess the maximum tolerated dose of the drug. The side effect of drug can also be evaluated in this phase. Most important thing is drug safety that can be evaluated in phase I. the dose of drug can be increased if it did not show any possible side effect, so that researchers can decide safest dose to therapy. Mainly healthy volunteer choose for study in experiment, in some cases the subject with disease may also required for the different kind of study.
Human participants- 20-100 Drug dose- Ascending dose Drug toxicity curve and dose efficacy curve obtained by this study. This includes single and multiple ascending doses. There are two types of escalation method one is rule based and another is model based. |
(Storer 1989; Jonas et al. 2007) | |||||
| Clinical trial phase | Description | Reference | |||||
| Phase II | Phases I and II are consider to be most perfect method to evaluate dose MSD that means, the maximum dose were drug is safest to administer and found to be non-toxic with desire therapeutic response. However phase I mainly target on MTD. Phase II trial help to assess the drug potential efficacy and therapeutic usefulness for the particular disease. Human participants:100-300,Study purpose- to assess how drug work and again there is evaluation of the safety of drug. Drug dose- therapeutic drug dose that find out during phase I is administered here. This study divided into two parts i.e., pilot clinical trial for efficacy and safety testing in diseased subject. Another one is rigorous trial made to demonstrate efficacy. The drug development step is terminated here if drug showing any toxic effects. This phase involve human subject showing many exclusive aspects than phase III. The randomized clinical trial design and the case studies are involved in this phase. The single stage and multi stage concepts are associated with phase II. The adaptive clinical trial is based on the accumulated data of interim that have been used for flexibility study and testing of efficacy. This is very helpful during trial to modify the design. | (Gehan 1961; Coffey et al. 2008; Brannath et al. 2010) | |||||
| Clinical trial phase | Description | Reference | |||||
| Phase III | This phase allow experimental clinical study with detail assessment of the treatment protocol by comparing data of new treatment with standard treatment. This is the most well known scientific investigation of the newer drug treatment. This phase is also known as pre- marketing phase. This phase consumes time and it is very expensive trial.
Human subject- 1000- 3000 This phase includes randomized controlled trials, uncontrolled trial, historical control, and no randomized concurrent trial etc. This trial is classified into two steps, one is trial after efficacy study but before the NDA submission, second one is carried out after the NDA submission but before the approval. In the year 1980 the FDA has given the guidance document which states that efficacy must be showed to prolongation of life and enhances the health related quality of patients life. |
(Pazdur 2008) | |||||
| Clinical trial phase | Description | Reference | |
| Phase IV | In this phase the treatment that proven to be safe, effective and good quality are made available to general public by authorization of FDA. The public, health professional people take the health benefits of marketed drug, but there are still some lacuna i.e., not all the safety and efficacy criteria has determined. There is need to continue the evaluation of safety on risk benefit ration, for this FDA requires to permit post marketing survey. This involves all the studies other than routine survey. This post marketing surveillance give actual mechanism of action of drug on given populations. In this drug related adverse effect can be observed. Long term adverse effect of the drug can be assessing in this phase, also the healthcare expenses and outcome can be determined. This phase IV result in drug can be removed from the market or may restrict to certain parameters. | (Committee on Ethical and Scientific Issues in Studying the Safety of Approval Drugs, 2012; Faden et al.2012). | |
Figure 1: diagrammatic representation of various laws, regulations and guidelines that
plan and monitor the clinical trial conducted in India.
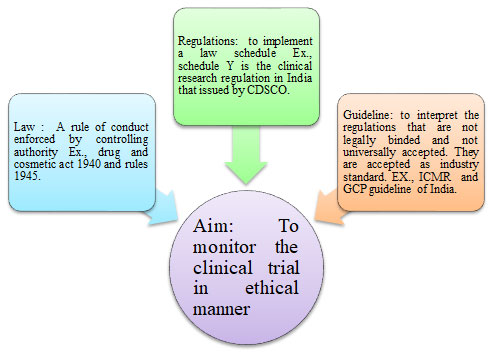
Figure 2: The figure showing criteria that should be meet before conducting actual clinical trial in India.

India has the second largest pharmaceutical market in Asia that a rate of US$ 5.40 billion and its growth rate is about 9% per year. There are around 1.2 billion people in India. And about 1.5 % of clinical trials were registered at the national institute of health, the USA from India in the year 2013. There is a wide scope of clinical research in India but with this, there is a need to follow international and national rules, regulations and guidelines that are associated with clinical research. A clinical trial should be conducted by adapting ICH –GCP rules in India (World Medical Association 2013; Saxena et al. 2014).
Good Clinical Practice is the international ethical standard for commencing biomedical research that involves the human subject in their experimental study. This standard ensures the rights, well-being and safety of the person who is participating in research work. This is not specific but it is general and can be applied to all the protocols (Saxena et al. 2014).
ICH in the technical requirement for human subject use in clinical research: This platform brings various regulatory authorities together, such as Europe, Japanese, and US authorities and also the pharmaceutical company expert to share scientific and technical aspects of the registration of products (Council for International Organization Of Medical Science 2002). US FDA adopts the code of GCP initially and that is applicable to all the sponsors and investigators. Other countries don’t have such a code, some country do not accept data from other countries. Europe and Japan have their own guideline that is somewhat similar to other guidelines but not exactly the same.
The person who is interested to market his product in the USA must go through their guidelines and submit clinical data. Canada, Australia, and Nordic countries formed a huge market that involves in ICH 1990, in this WHO is the facilitator and the IFPMA (International Federation of Pharmaceutical Manufacturers Associations). The ICH guidelines were completed in the year 1996 and this involves the criteria of safety, quality and efficacy with multidisciplinary. Good clinical practice involves in the E6 chapter. This helps in analysis, to guide technical issues and provides sufficient knowledge to register so that repetition of tests or studies can be avoided. The ICH- GCP provides the responsibilities to the ethics committee, sponsors and an investigator, along with this also gives a brief idea about protocol requirements and investigator brochure (IB) and trial document (Saxena et al. 2014).
Ethics committee: This committee has a role in developing a constitution and standard operating procedures that must involve the members and the appointment conditions. The ethics committee looks after all the protocols of the research study, the informed consent form of the patient and other necessary documents that are associated with the research proposal. After a detail reviewing of all document’s ethics committee supposed to give approval to the study and supervise the whole study (Desai et al. 2013).
It also provides a safeguard to the subject who is involved in the study, the well-being of the subject is the priority of the committee. The committee must have to overlook the experimental study on daily basis. The committee must have to maintain confidentiality and also maintain all the records and documental proof. All records should be maintained until 5 years of duration after completion of the study. This committee also has a role in examining SAE reports and sending them to DCGI. This committee allows DCGI to inspect and follow all national and international protocols (Bonthagarala et al. 2017).
Sponsors: A sponsor is the one who takes the responsibility for starting, managing and giving financial support to a clinical trial. The investigator can also play the role of a sponsor performing all the duties related to the trial called the role of sponsor. Sponsors have the right to give clinical trial-related activities to any scientific body or contract research organization. Such transfer should be documented well in writing format. The sponsor should evaluate the investigator before starting the actual study, the sponsor must be a trained person, and having an experience and knowledge in conducting the trial.
The sponsor has the responsibility to assess the recruiting potential of the investigator based on previous reports, space, time, equipment, teamwork, lab facility etc. the investigator brochure must be provided to the investigator before commencing to study. Another responsibility of the sponsor is to obtain approval from DCGI and Institutional Ethical committee. The serious adverse event that happens during the trial should be recorded within the given period. The sponsor has to submit the protocol report, case report form and ICF. They have to monitor, assist, and evaluate all the data and financing (Council for International Organization Of Medical Science 2002; Bonthagarala et al. 2017).
Investigator: The actual in charge of all the studies conducted and supervision of this whole procedure is done by the investigator. The investigator should possess the criteria to fulfil his duty which include qualification first, then training experience and treatment facility related to the protocol of the study. Before commencing to trial, the sponsor and investigator must have signed an agreement on paper, on protocol, monitoring responsibilities and trial-related duties etc. The investigator has the job to obtain approval from the institutional ethics committee, to get ICF from each human subject involved in the study. Any serious adverse event that occurs during the trial, needs to be reported as early as possible in the fixed period. Investigator does not take more than three trials at the time (Council for International Organization Of Medical Science 2002; Saxena et al. 2014).
What is CTRI? (The Clinical Trials Registry-India) and need of it: The CTRI were launched on 20/7/2007, this is the absolutely free, searchable online site where clinical trial registration can be done. On this platform, all types of clinical trial-related studies can be registered. The PG theses can also be registered here. Recently, there are many types of registration that can be done on CTRI Like- intervention trials, observational data, bioavailability, and also the phase IV studies. All these trials are easy to search and handle by common people from its official home page (Adhikari et al. 2018).
Medical science has developed so fast because of which there is an increase in new therapeutic measures and ultimately newer methods for designing the fresh agents. The trials that are not ethically conducted further lead to withdrawal from the market in future, therefore it is essential to create transparency and easy watch out on clinical trials for research persons and the public (Adhikari et al. 2018). In the year 2008, the editors of the biomedical journal support clinical trial registration. They had published a joint statement that states “don’t accept the unregistered clinical trial for publication in any journal from the year 2010” (Bavdekar et al. 2008; Adhikari et al. 2018).
The World Health Organization (WHO) recognize the CTRI as the primary registry in the year 2008, from this year every month data gets transferred from CTRI to the international clinical trials registry site. This is the medium where globally all clinical trial gets registered. In the year 2009, the CDSCO made it compulsory for every clinical trial that is supposed to be conducted, to do their registration in CTRI first (Adhikari et al. 2018).
To maintain a smooth flow in trial registration the CTRI software was upgraded and revised all the data on dated 15th march 2011. This new software made it possible to help the overall system go paperless and there is also a facility to upload all regulatory approvals on an online basis. Furthermore, the CTRI arrange a number of workshops that involve ethics committee members, to grow the trial registration and to make awareness among people. Various ethics committees made it mandatory for trial registration. The improvement was observed when AYUSH also come to accept the trial registry. The E- utorial was launched in 2015. This tutorial is made to guide the process of trial registration (Adhikari et al. 2018).
The importance of CTRI in the registration of PG theses is CTRI registration is important in the registration of PG theses that would be helpful in raising the standard of research so that there will be a limit to the repetition of already done research work. This can act as ready guide that answers the research questions like how one can start research work, how one can start clinical research, student can receive information regarding protocols and strategies of research work that would be beneficial in their future work (Adhikari et al. 2018).
The future expectation of CTRI: The registration of the trial is not enough to fulfil all criteria, there should be the disclosure of the result is also mandatory to maintain transparency and accessibility. Because of this reason, ICMR joins the WHO on 18th may 2017 to make sure that there is a disclosure of results within one year after completion of the trial. Taking this into consideration the CTRI develop the structural framework that should involve the patient number, characterization of baseline, primary and secondary outcome, and adverse event of the drug. This protocol makes it easy to reach the public hand. Since 1st April 2018, CTRI moved towards prospective trial registration that is only for clinical studies. This is applicable to all types of clinical studies that are submitted for registration (Adhikari et al. 2018).
CONCLUSION
The overall conclusion of the present article found that it is essential to describe the clinical research, how it was conducted and what clinical trial phases are. To understand the drug therapy that is required to pass through multiple phases before coming to market and the importance of safety and health of a population. The various regulatory bodies that ensure all experimental studies should be conducted ethically. It is essential to maintain the highest quality and standard in drug safety and efficacy evaluation parameters as it directly links with wellbeing of the population.
Conflict of interests: Authors declare no conflict of interest to disclose
Funding source: There is no source of funding.
REFERENCES
Adhikari T, Gupta J, Rao MVV, Juneja A et al. (2018). Clinical Trials Registry-India: an overview and new developments. Indian Journal of Pharmacology, 50(4), Pages 208.
Ambroz A, Blackburn B, Chalmers T et al. (1981). A method for assessing the quality of a randomized control trial. Controlled Clinical Trials, 2(1), Pages 31-49.
Avins, A. (1998). Can unequal be more fair? Ethics, subject allocation, and randomised clinical trials. Journal of Medical Ethics, 24(6), Pages 401-408.
Bavdekar S.B, Gulati R.K, Parikh P, et al. (2008). Statement on publishing clinical trials in India biomedical journals. Indian Journal of Ophthalmology, 56(3), Pages 177.
Bonthagarala B, Evangeline L, Mounica N.V.N, et al. (2017). Regulatory process and ethics for clinical trials in India (CDSCO). The Pharma Innovation, 6(4, Part C), Pages 165.
Brannath W, Burger H, Glimm E, et al. (2010). Comments on the Draft Guidance on Adaptive Design Clinical Trials for Drugs and Biologics of the U.S. Food and Drug Administration. Journal of Biopharmaceutical Statistics, 20(6), Pages 1125-1131.
Browne, L. and Graham, P. (2014). Good intentions and ICH-GCP: Trial conduct training needs to go beyond the ICH-GCP document and include the intention-to-treat principle. Clinical Trials, 11(6), Pages 629-634.
Coffey C. and Kairalla J. (2008). Adaptive Clinical Trials. Drugs in R & D, 9(4), Pages 229-242.
Collins J, Doroshow J, Helman, L. et al. (2007). Compressing drug development timelines in oncology using phase ‘0’ trials. Nature Reviews Cancer, 7(2), Pages 131-139.
Committee on Ethical and Scientific Issues in Studying the Safety of Approved Drugs, Board on Population Health and Public Health Practice and Institute of Medicine, (2012). Ethical and scientific issues in studying the safety of approved drugs. National Academies Press.
Council for International Organizations Of Medical Science (2002). International ethical guidelines for biomedical research involving human subjects. Bulletin of medical ethics, (182), Pages 17-23.
DeMets D.L, Friedman L.M, Furberg C.D., et al. (2015). Fundamentals of clinical trials. springer.
Desai C, and Pandya M. (2013). Compensation in clinical research: The debate continues. Perspectives in clinical research, 4(1), Pages 70.
Esource.com. (2022). Behavioral programs | E Source. [online] Available at: <https://www.esource.com/topics-category/behavioral-programs> [Accessed 17 March 2022].
Faden R.R, Goodman S.N and Mello M.M (2012). Ethical considerations in studying drug safety—the Institute of Medicine report. New England Journal of Medicine, 367(10), Pages 959-964.
FDA (2022). U.S. Food and Drug Administration. [cited 23 March 2022]. Available from: https://www.fda.gov/patients/drug-development-process/step-3-clinical-research
Gehan E. (1961). The determination of the number of patients required in a preliminary and a follow-up trial of a new chemotherapeutic agent. Journal of Chronic Diseases, 13(4), Pages 346-353.
Haahr, M. and Hróbjartsson, A. (2006). Who is blinded in randomized clinical trials? A study of 200 trials and a survey of authors. Clinical Trials, 3(4), Pages 360-365.
Jonas W, Khuri F, Porter A. et al. (2007). Translation of Innovative Designs Into Phase I Trials. Journal of Clinical Oncology, 25(31), Pages 4982-4986.rics, 45(3), Pages 925.
Le Tourneau C., Lee J. and Siu L. (2009). Dose Escalation Methods in Phase I Cancer Clinical Trials. JNCI: Journal of the National Cancer Institute, 101(10), Pages 708-720.
Mahan V. (2014). Clinical Trial Phases. International Journal of Clinical Medicine, 05(21), pages 1374-1383.
Marchetti, S. and Schellens, J. (2007). The impact of FDA and EMEA guidelines on drug development in relation to Phase 0 trials. British Journal of Cancer, 97(5), Pages 577-581.
Pazdur, R. (2008). Endpoints for Assessing Drug Activity in Clinical Trials. The Oncologist, 13(S2), Pages 19-21.
Saxena, P. and Saxena, R. (2014). Clinical trials: changing regulations in India. Indian journal of community medicine: official publication of Indian Association of Preventive & Social Medicine, 39(4), Pages 197.
Storer B. (1989). Design and Analysis of Phase I Clinical Trials. Biometrics, pp.925-937.
World Medical Association (2013). World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. Jama, 310(20), Pages 2191-2194.

