1School of Sciences, Department of Botany, Maharani Cluster University, Bengaluru – 560001, Karnataka, India.
2School of Sciences, Department of Chemistry & Biochemistry, Maharani Cluster University, Bengaluru – 560001,
Karnataka, India.
3Department of Biochemistry, Bangalore University, Jnanabharathi Campus, Bangalore-560056 India
4Post Graduate Department of Life Sciences, Mount Carmel College, (Autonomous) Bangalore-560052
Corresponding author email: nageshbabur@gmail.com
Article Publishing History
Received: 13/07/2020
Accepted After Revision: 24/09/2020
MicroRNAs (miRNAs) are small non-coding RNAs which regulate gene expression by cleavage or repression of target genes at post-transcriptional level by translational inhibition/ mRNA degradation. Niger (Guizotia abyssinica) is an important oilseed crop widely grown in India. Identification and expression of non-coding RNAs during abiotic remains unclear till date. Small RNA library was constructed by high throughput sequencing from control and stress tissues. Target genes of identified miRNAs were predicted using psRNA Target and their GO terms were annotated. The results were validated using RT-qPCR. In this study, we constructed the RNA libraries using next generation sequencing and 125 candidate miRNAs associated to high temperature stress were identified. The qPCR revealed miR395, miR396, miR319 were up-regulated by >15 folds. Most of the targets identified were transcription factors (SPL, MYB, GRF, NAC and GRAS) and oxidative stress. This is to our knowledge the first report for identifying the high temperature stress responsive miRNAs in Niger. Further, characterization and functional annotations of the target genes would provide insights into the regulatory mechanism employed to sustain extreme temperature.
Abiotic stress; Growth Factors; High throughput sequencing; Transcription Factors
Prathiba K. Y, Usha S, Suchithra B, Jyothi M. N, Ulfath T. K. S, Sharadamma N, Hoor F. S, . Nageshbabu R. Analysing the Response of Non-Coding RNA of Niger, Guizotia abyssinica Towards High Temperature and Associated Functional Predictions. Biosc.Biotech.Res.Comm. 2020;13(3).
Prathiba K. Y, Usha S, Suchithra B, Jyothi M. N, Ulfath T. K. S, Sharadamma N, Hoor F. S, . Nageshbabu R. Analysing the Response of Non-Coding RNA of Niger, Guizotia abyssinica Towards High Temperature and Associated Functional Predictions. Biosc.Biotech.Res.Comm. 2020;13(3). Available from: https://bit.ly/3gVHlRn
Copyright © Prathiba et al., This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) https://creativecommons.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
Niger (Guizotia abyssinica) is an important but neglected edible oil seed crop widely grown in India. Niger is grown in an area of 2.53 lakh hectares with the production of 0.83 lakh tonnes and the productivity of 326 kg/hectare (Dugas and Bartel 2004). The crop of dry areas grown mostly by tribal and desired attention was not accorded on the biotic and abiotic stress conditions. Being a rain fed crop, Niger is exposed many abiotic stresses like drought, high temperature, salt and low nutrients which adversely affect the plant productivity. To date, the reports pertaining to biochemical effects of high temperature or role of miRNAs under high temperature on Niger cultivar is sparse. Thus, it is necessary to elucidate stress tolerance mechanisms by the involvement of miRNAs during high temperature to develop/improve the tolerance cultivar. Small RNAs have emerged as ubiquitous key molecules regulating gene expressionat the post-transcriptional level, either by repressing mRNA translation or mediating the degradation of the targeted mRNAs depending on their degree of complementarity (Suryanarayana et al., 2018).
Precursor stem-loop secondary structures are characteristic features of miRNAs and are conserved across species (Carrington and Ambros 2003; Bartel and Sunker 2005). In plants, small RNAs and more specifically, microRNA (miRNA)s (~21 nt), have been functionally associated with development, biotic and abiotic stress (Lelandais-Briere et al., 2009). Regulation of miRNAs by abiotic stresses was initially reported independently by (Jones-Rhoades et al., 2006) and (Sunkar et al., 2004). Subsequently, a number of reports have been published which echoed that miRNAs are themselves regulated by abiotic factors and they in turn, control the levels of target genes involved in governing the stress responses. Two of the most featuring examples are miR398 and miR395, which have been repeatedly shown by independent groups to regulate cellular responses in many different stresses (Li et al., 2010; Khraiwesh et al., 2012).
Bharadwaj et al. (2014) had identified 15 conserved miRNAs in heat stressed Brasicca libraries and validated the expression of miR395 which is indented as another miRNA involved in heat stress response other than miR398 as established in Arabidopsis (Lu et al., 2013) and French bean (Naya et al., 2014). Recently, Kavya and Devraj (2020) have reported the up-regulation of miR166a, miR156, miR6173, miR169e-5p, miR6478 and miR166U under salinity stress. However, till date no reports of miRNA characterization in Niger drawn us towards elucidating the role of in adaptive strategies employed by the oil seed plant to overcome the climatic cues. In this view, we ensued with heat treating the plants at 48 ºC for 8 h and profiled their small RNA expression using high throughput sequencing and validating the results with qPCR. We identified 125 conserved miRNAs belonging to 45 families. The cumulative studies of relative quantification using RT-qPCR. Our results emphasize that differential expression would render stress tolerance and has important implications for gene regulation under abiotic stress conditions, (Kavya and Devaraj, 2020).
MATERIAL AND METHODS
Plant material and high temperature stress treatment: Niger seeds were surface sterilized and grown under controlled conditions at 28 °C day/25 °C night with 12 h light/12 h dark photo period. After 6 day of germination, seedlings were exposed to high temperature stress (42 °C for 1 h (induction); 45 °C for 1 h and 48 °C for 6 h). Tissues (shoot) were harvested immediately and stored at -80 °C for further analysis.
Small RNA library Construction and sequencing: Following RNA extraction, small RNA library (control and stress) was prepared according to the True Seq small RNA sample prep Kits protocol (Illumina San Deigo USA). The quality and quantity of total RNA were analyzed using Agilent 2100 bio-analyzer. Ten to thirty nt sRNAs were purified from 15% denaturing polyacrylamide gel and then ligated with the 5’ and 3’ adapters. After being reverse transcribed by Superscript II reverse transcriptase (Invitrogen, USA), sRNAs were amplified by PCR. High throughput sequencing was performed using Nextseq500 platform (Illumina, USA).
Identification of miRNAs, target predictions and GO analysis: After Illumina sequencing, high quality small RNA reads were extracted from raw reads through filtering the adapter dimers and low-quality tags. Subsequently, unique sequences with 18~25 nucleotides length were mapped with ESTs of Niger precursors in miRBase 21.0 (http://www.mirbase.org/) by BLAST search to identify conserved and novel miRNAs. The potential candidate miRNAs were identified by folding the flanking EST sequence of unique small RNAs using mfold web server (Zuker et al., 2004). Parameters were set based on the criteria for annotation of plant miRNAs by Meyers et al (2008). To identify novel miRNAs, the miRDeep -P program was used to obtain all candidate precursors with hairpin-like structures that were perfectly mapped by sequencing tags. Target predictions were performed using the psRNATarget web server with ESTs of Niger (http://plantgrn.noble.org/ psRNATarget/analysis) using default parameters with a maximum of 3 expectation cut-off. The GO terms of the target genes were annotated according to their biological process, molecular functions, or involvement as cellular components using Blast2GO. The enzyme mapping of the annotated sequences was performed directly using the GO terms and KEGG orthologs.
qRT-PCR analysis of miRNA: RT-qPCR was used to validate the results obtained from the high throughput sequencing of miRNAs. RNA was isolated using Trizol (Invitrogen) as per manufacturer’s instruction. 1 µg Total RNA was reverse transcribed using stem-loop primers designed according to Chen 2005 and gene specific primers for target genes using One Step Prime Script miRNA cDNA Synthesis Kit (Takara, Japan). Rt-qPCR was performed using SYBR premix ExTaq (Takara, Japan) and all the primers used were listed in Supplementary file 1. Small nuclear RNA U6 and GAPDH were used as internal controls to normalize the miRNA expression and target genes expression, respectively. Subsequently, the quantification was carried out using (CFX-96, Bio Rad). Three biological replicates were used per sample in addition to technical replicates along with a no template control and no RT-enzyme control. The data were analyzed using 2-ΔΔCT method and reported as means ± standard errors (SE) of three biological replicates. Fold changes were determined by using the ratio of normalized expression of stress against control samples and represented as log 2 values.
Small RNA libraries from stress and control seedlings were screened using Nextseq 500 (Illumina Inc, USA) generated nearly twenty million total raw reads. After removing low-quality sequences, adapters, and small sequences (< 17 nt long), 18,445,935 and 19,445,620 representing high quality sequences were obtained from stress and control libraries respectively. Further to determine the stress specific miRNAs, the sequences were filtered against the control library. Only reads found in stress library were considered for small RNA identification. An in-house database comprising of non-coding RNAs expect miRNAs from Rfam (12.0) was created and used to filter small RNA fragments corresponding to non-coding RNAs such as tRNAs, rRNA, sncRNAs etc., The small RNA length distribution (16-30 nt) of each library showed that the most abundant and diverse species were those 21-24 nt in length, a typical size range for Dicer-derived products (Fig1) and termed as unique sequences which were further considered for identification of conserved miRNAs. In order to identify the conserved miRNAs, the unique sequences were mapped against mature miRNAs in miRBase (v 21). Following Blastn searches and further sequence analysis, a total of 125 miRNAs belonging to 45 families were identified. miR156, miR399 and miR169(Fig 1) represents the most abundant miRNA family (Fig 2).
Figure 1: Data filtering and Length distribution. A. Pie plot of data filtering B. Sequence read length distribution of mappable high temperature stress responsive small RNAs (sRNAs).
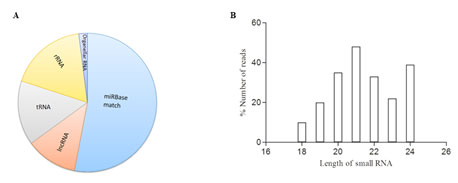
Figure 2: Expression levels of known miRNA families. The expression levels of the miRNA families were normalized by the total number of reads in each of the respective libraries.
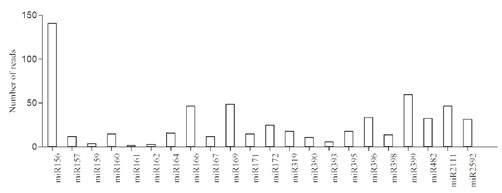
All the foresaid conserved miRNA families, possessed miRNA* sequences, identified with their stem-loop structures (Supplementary file of structures) prompts as additional evidence in support of the authenticity of the miRNAs. Since, the precursors and mature miRNAs are highly conserved in plants, it is feasible to define the MFEI values of newly identified conserved miRNAs based on their homologs used as reference sequences. To elucidate the biological functions of high temperature stress associated miRNAs were assessed using the psRNA Target software. A total of 750 potential target genes were identified, based on their perfect or near-perfect complementarity to their target mRNA sequences. Most of the predicted targets were extensively involved in different biological process involving a large number of gene families. Some of these encoded transcription factors, DNA replication proteins, and those involved in cellular metabolism in addition to wide range of stress response associated proteins. Detailed annotations of these results are presented in (Supplementary File 4). miR156 and miR157 targets SPL protein and miR166 targets Homeobox-leucine zipper family protein respectively.
Figure 3: Gene ontology of differentially expressed miRNA targets.
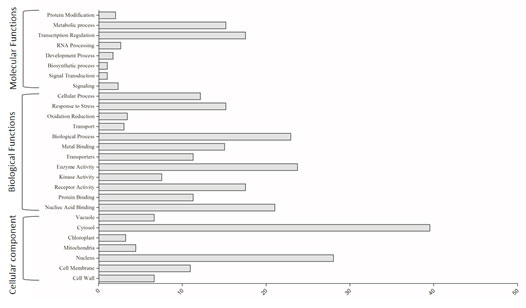
The GO terms of the targets were annotated according to their biological function and highest percentage of genes falls in to cellular components. The enzyme mapping of the annotated sequences was performed using direct GO for the enzyme mapping and the Kyoto Encyclopedia of Genes and Genomes (KEGG) for the definitions of the KEGG orthologs. The miRNA targeted genes belonged to various biological process, cellular components and molecular functions as depicted in (Fig 3). The maximum numbers of target genes were involved in biological process, including both metabolic and cellular process. However, target genes in binding were the most abundant (80%) within the molecular function category. We have also found many genes representing transporters, receptors and signalling molecules including kinases. The compartmentalization of target genes revealed most of them are membrane proteins and localized in nucleus and cytoplasm.
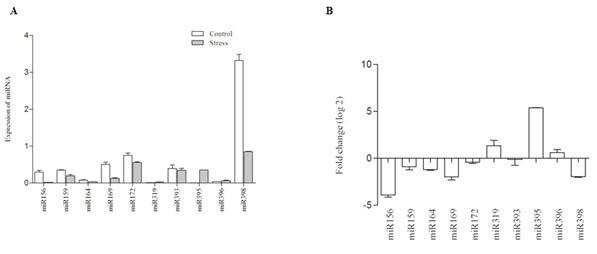
Figure 4: A. Quantification of miRNA abundance via RT-qPCR Differential expression of 10 miRNAs following high temperature stress. B. Fold changes (log2) in known miRNA levels in response to high temperature stress.
The putative miRNA targets in Niger were predicted using the psRNATarget program. The target genes (approximately 750 different transcripts) were extensively involved in different biological processes involving a large number of gene families. Some of these genes encoded transcription factors, DNA replication proteins and those that are involved in cellular metabolism in addition to a variety of stress response-associated proteins. miR156, miR166 and miR319 target genes encode Squamosa promoter-binding protein, Homeobox-leucine zipper protein and MYB domain proteins respectively, as previously reported (Ferdous et al. 2015).
SPL genes forms one of the most targeted gene and we found 09 SPL genes belonging to Clade-I as major targets from the family. SPLI proteins constitute diverse family of transcription factors which are crucial in plant growth and development. Many studies established the role of SPL proteins in transition of juvenile to adult phase, reproductive transition, trichome development, apical dominance, inflorescence branching, fruit ripening, pollen sac development, and copper homeostasis (Unte et al., 2003; Manning et al., 2006; Wu and Poethig, 2006; Schwarz et al., 2008; Wang et al., 2009; Yamaguchi et al., 2009; Yamasaki et al., 2009; Jiao et al., 2010; Miura et al., 2010; Preston and Hileman, 2010; Yu et al., 2010).
Previous studies established the involvement of miR156, miR157 and miR398 in plant stress response mechanism, specifically in maintaining redox equilibrium under heat stress and copper homeostasis (Lu et al 2012; Preston and Hileman 2013). They demonstrated that, the repression of miR398b and miR398c in Arabidopsis would result in loss of degradation of CSD1 and CSD2, which would support channeling of limited copper to support photosynthetic activity under high temperature induced oxidative stress and copper deficiency. They also showed that the repression of miR156 and miR157 would lead to activation of SPLII genes involved in copper independent photosynthetic pathways. The other major transcription factor targeted by conserved miRNAs is found to be MYB domain proteins.
Figure 5: A. Determination of miRNA target gene expression via RT-qPCR analysis of Niger whole Control and heat stressed seedlings B. Fold changes (log2) 10 selected miRNAs and their targets determined by RT-qPCR.

MYB is one among the vital proteins in plants which involved in development, metabolism, hormone signal transduction, disease resistance and environmental cues (Jagadeeswaran et al., 2009). AtMYB44 was demonstrated to act at front line of salt stress induced cellular re-programming and suggested AtMYB44 prevent stress triggered tissue collapse by rapid removal of ROS (Pashkovskiy and Ryazansky 2013). The most widely targeted class of genes is encoding Toll/Interleukin-1 receptor-nucleotide binding site-leucine-rich repeats (TIR-NBS-LRR). Members of the TIR-NBS-LRR gene family are established as genuine targets for miR2118. Nucleotide Binding Site -Leucine Rich Repeats (NBS-LRR) gene products confers disease resistant in plants and found to harbor secondary small RNAs (tasiRNAs). The miR482 mediated gene cleavage would activate a cascade of defense reactions upon pathogen attack. In cotton, it is demonstrated that, the expression of NBS-LRR was induced to promote the generation of tasiRNAs which mediate small RNA mediated gene silencing, a probable mechanism of plant defence against viral and fungal attack. miR482 and miR2118 were clustered in Medicago trancatula and targets NBS-LRR disease resistant gene (Persak and Pitzschke 2014).
We also observed 10 ESTs coding GRF proteins targeted by miR396. miR396, which targets growth regulating factors (GRFs) are an important class of transcriptional regulators involved in the control of cell proliferation in leaves (Palatnik et al., 2007). RT-qPCR profile showed strong up-regulation of miR396 (1-fold) which appears to play a negative role in cell proliferation due to reduced cell division and it is reasonable to hypothesize that, as in Arabidopsis, miR396 restricts GRF expression, contributing to proliferation arrest in expanding cells (Rodruiguez et al., 2010). Recent studies imply an intriguing regulatory network between miR164, miR396, and miR319 in Arabidopsis: miR319 regulates five members of the TCP transcription factor family (TCP2, 3, 4, 10, 24) that inhibits cell proliferation and directly bind the miR164 promoter region (Khraiwesh et al., 2010; Martin-Trillo and Cubas 2010). Total RNA from the tissues of control and stressed Niger plants were used to validate the miRNAs. The total RNA was converted to cDNA using stem – loop primers. The expression of 10 conserved miRNAs were considered for the study based on their read count/abundancy using qRT-PCR (Martin-Trillo and Cubas 2010).
The expression levels of the Niger miRNAs under high temperature were significantly altered compared with those of the control seedlings. Based on the sequencing data, we selected 10 miRNAs, which were also reported to as stress responsive miRNAs in other species. Of the 10 miRNA we found only 3 of them were found up-regulated. miR395 was found to be highly responsive and was induced by 12 folds when compared with the control seedlings. miR319 and miR396 were up-regulated by 2-folds. Among the down regulated miRNAs, 8-fold depression was observed with miR156, 4-fold repression was found with miR169 and miR398, and an average of 2-fold repression was observed with other miRNAs (Fig 4). To validate the expression of targets the expression analysis was carried out with selected conserved targets of miRNAs. Since miRNAs were conserved across the kingdom, the genes targeted is also conserved with few exceptions. Since Niger lack complete genome data, we selected conserved targets, for the analysis. The expression profile substantiated the previous observations of negative correlation with their respective miRNAs. ATP sulpharylase was highly repressed by 14 folds, followed by DCL1 and GRF3. DUO1 and SPL7 were induced by 11 and 9 folds (Tian et al., 2014).
However, the target genes exhibited marginal changes in their expression. This may be due to the involvement of transcription regulatory factors other than miRNAs whose expression may not alter due to stress induction (Fig 5). In the present study, RT-qPCR was carried out to study the expression of randomly selected conserved miRNAs representing the most stress responsive miRNA families. All the miRNAs showed sensitivity towards high temperature and our results evidences the miRNA abundance and their expression trends which is consistent with the previous results. We also observed induction of miR395, miR166/167 by 2-fold repression of miR156, miR171 which discern the effects of high temperature on Niger. Many miRNAs were temperature sensitive, for instance, miR160, miR166, miR167 and miR393 were up-regulated in barley and wheat upon heat treatment. Differential expression trends of miR156, miR159, miR396 and miR398 were also observed in Arabidopsis (Jagadeeshwaran et al., 2009) and Broccoli (Tian et al., 2014). miR398 was the most extensively studied miRNA with respect to heat stress. It is demonstrated that the repression of the miR398 under high temperature could render plant tolerant to heat induced oxidative damages (Naya et al., 2014, Lu et al., 2012 and Yu et al., 2012).
CONCLUSIONS
We have identified many non-coding RNAs involved in high temperature stress in Niger were discovered by high-throughput sequencing and annotated their targets. Further, the role of miRNAs target interaction, GO analysis and protein interaction of target gene were studied which showed the identified miRNAs paly an important role in cellular homeostasis in addition to growth and development. However, the detailed mechanism of miRNAs under high temperature stress still requires detailed characterization. However, these finding will contribute for further investigations of miRNAs in Niger under abiotic stress conditions.
ACKNOWLEDGMENTS
The authors would like to thank DST-FIST, UGC-CPE and DBT Star College Scheme for Instrumentation facility and consumables.
Conflict of interest: The authors declare no conflict of interest.
REFERENCES:
Arenas-Huertero, C., Pérez, B., Rabanal, F., Blanco-Melo, D., De la Rosa, C., Estrada-Navarrete, G., and Reyes, J. L. (2009). Conserved and novel miRNAs in the legume Phaseolus vulgaris in response to stress. Plant molecular biology, 70(4), 385-401.
Barozai, M. Y. K., Irfan, M., Yousaf, R., Ali, I., Qaisar, U., Maqbool, A., and Riazuddin, S. (2008). Identification of micro-RNAs in cotton. Plant Physiology and Biochemistry, 46(8-9), 739-751.
Barrera-Figueroa, B. E., Gao, L., Diop, N. N., Wu, Z., Ehlers, J. D., Roberts, P. A., and Liu, R. (2011). Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC plant biology, 11(1), 127.1-17.
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell, 136(2), 215-233.
Bita, C., and Gerats, T. (2013). Plant tolerance to high temperature in a changing environment: scientific fundamentals and production of heat stress-tolerant crops. Frontiers in plant science, 4, 273.
Ding, D., Zhang, L., Wang, H., Liu, Z., Zhang, Z., and Zheng, Y. (2009). Differential expression of miRNAs in response to salt stress in maize roots. Annals of botany, 103(1), 29-38.
Dugas, D. V., and Bartel, B. (2004). MicroRNA regulation of gene expression in plants. Current opinion in plant biology, 7(5), 512-520.
Fahlgren, N., Howell, M. D., Kasschau, K. D., Chapman, E. J., Sullivan, C. M., Cumbie, J. S., and Carrington, J. C. (2007). High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PloS one, 2(2), e219.
Fujii, H., Chiou, T. J., Lin, S. I., Aung, K., and Zhu, J. K. (2005). A miRNA involved in phosphate-starvation response in Arabidopsis. Current Biology, 15(22), 2038-2043.
Griffiths-Jones, S., Grocock, R. J., Van Dongen, S., Bateman, A., and Enright, A. J. (2006). miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids research, 34(suppl_1), D140-D144.
Griffiths-Jones, S., Grocock, R. J., Van Dongen, S., Bateman, A., and Enright, A. J. (2006). miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids research, 34(suppl_1), D140-D144.
Jia, X., Wang, W. X., Ren, L., Chen, Q. J., Mendu, V., Willcut, B., and Tang, G. (2009). Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant molecular biology, 71(1-2), 51-59.
Kavya, N. H., and Devaraj, V. R. (2020). Genomic of salt induced microRNAs in Niger (Guizotia abyssinica Cass.) Plant gene. doi.org/10.1016/j.plgene.2020.100242
Kotak, S., Larkindale, J., Lee, U., von Koskull-Döring, P., Vierling, E., and Scharf, K. D. (2007). Complexity of the heat stress response in plants. Current opinion in plant biology, 10(3), 310-316.
Liang, G., He, H., and Yu, D. (2012). Identification of nitrogen starvation-responsive microRNAs in Arabidopsis thaliana. PloS one, 7(11), e48951.
Liu, Y., Ji, X., Zheng, L., Nie, X., and Wang, Y. (2013). Microarray analysis of transcriptional responses to abscisic acid and salt stress in Arabidopsis thaliana. International Journal of Molecular Sciences, 14(5), 9979-9998.
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., and Griffiths-Jones, S. (2008). Criteria for annotation of plant MicroRNAs. The Plant Cell, 20(12), 3186-3190.
Moxon, S., Jing, R., Szittya, G., Schwach, F., Pilcher, R. L. R., Moulton, V., and Dalmay, T. (2008). Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome research, 18(10), 1602-1609.
Nageshbabu, R., Jyothi, M. N., and Sharadamma, N. (2013). Expression of miRNAs regulates growth and development of French bean (Phaseolus vulgaris) under salt and drought stress conditions. ISCA Journal of Biological Sciences, 2(1), 52-56.
Nageshbabu. R., Jyothi, M.N., Usha, Sharadamma N., Rai D. V and Devaraj V. R. (2014) Identification of miRNAs from French bean (Phaseolus vulgaris) under low nitrate stress. Turk J Biochem, 39 (1),1-8.
Naya, L., Paul, S., Valdés-López, O., Mendoza-Soto, A. B., Nova-Franco, B., Sosa-Valencia, G., and Hernández, G. (2014). Regulation of copper homeostasis and biotic interactions by microRNA 398b in common bean. PLoS One, 9(1), e84416.
Pashkovskiy, P. P., and Ryazansky, S. S. (2013). Biogenesis, evolution, and functions of plant microRNAs. Biochemistry (Moscow), 78(6), 627-637.
Shen, J., Xie, K., and Xiong, L. (2010). Global expression profiling of rice microRNAs by one-tube stem-loop reverse transcription quantitative PCR revealed important roles of microRNAs in abiotic stress responses. Molecular Genetics and Genomics, 284(6), 477-488.
Shinozaki, K., and Yamaguchi-Shinozaki, K. (2007). Gene networks involved in drought stress response and tolerance. Journal of experimental botany, 58(2), 221-227.
Sunkar, R., Chinnusamy, V., Zhu, J., and Zhu, J. K. (2007). Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends in plant science, 12(7), 301-309.
Sunkar, R., Kapoor, A., and Zhu, J. K. (2006). Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. The Plant Cell, 18(8), 2051-2065.
Sunkar, R., Zhou, X., Zheng, Y., Zhang, W., and Zhu, J. K. (2008). Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC plant biology, 8(1), 25.
Suryanarayana, L., Sekhar, D., and Tejeswara Rao, K. (2018). Genetic divergence studies in Niger (Guizotia abyssinica L.) genotypes. Journal of Pharmacognosy and Phytochemistry, 7(5), 725-727.
Tagami, Y., Inaba, N., Kutsuna, N., Kurihara, Y., and Watanabe, Y. (2007). Specific enrichment of miRNAs in Arabidopsis thaliana infected with Tobacco mosaic virus. DNA research, 14(5), 227-233.
Trindade, I., Capitão, C., Dalmay, T., Fevereiro, M. P., and Dos Santos, D. M. (2010). miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta, 231(3), 705-716.
Tuskan, G. A., DiFazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., and Schein, J. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science, 313(5793), 1596-1604.
Valdés L., O.., Huertero, C. A., Ramirez, M., Girard, L., Sanchez, F., Vance, C. P., and Hernandez, G. (2008). Essential role of MYB transcription factor: PvPHR1 and microRNA: PvmiR399 in phosphorus deficiency signalling in common bean roots. Plant, Cell and Environment, 31(12), 1834-1843.
Valdés L, O., Yang, S. S., Aparicio F. R., Graham, P. H., Reyes, J. L., Vance, C. P., and Hernández, G. (2010). MicroRNA expression profile in common bean (Phaseolus vulgaris) under nutrient deficiency stresses and manganese toxicity. New Phytologist, 187(3), 805-818.
Wang, Q. L., and Li, Z. H. (2007). The functions of microRNAs in plants. Front Biosci, 12, 3975-3982.
Xin, M., Wang, Y., Yao, Y., Xie, C., Peng, H., Ni, Z., and Sun, Q. (2010). Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC plant biology, 10(1), 1-11.
Yu, X., Wang, H., Lu, Y., de Ruiter, M., Cariaso, M., Prins, M., and He, Y. (2012). Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. Journal of Experimental Botany, 63(2), 1025-1038.
Zhang, N., Yang, J., Wang, Z., Wen, Y., Wang, J., He, W., and Wang, D. (2014). Identification of novel and conserved microRNAs related to drought stress in potato by deep sequencing. PloS one, 9(4), e95489.
Zhang, Y. (2005). miRU: an automated plant miRNA target prediction server. Nucleic acids research, 33(suppl_2), W701-W704.
Zhou, Z. S., Wang, S. J., and Yang, Z. M. (2008). Biological detection and analysis of mercury toxicity to alfalfa (Medicago sativa) plants. Chemosphere, 70(8), 1500-1509.
Zhou, Z. S., Zeng, H. Q., Liu, Z. P., and Yang, Z. M. (2012). Genome wide identification of Medicago truncatula microRNAs and their targets reveals their differential regulation by heavy metal. Plant, cell and environment, 35(1), 86-99.
Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research, 31(13), 3406-3415.

