Department of Biological Science, Faculty of Science, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia
Corresponding author email: ahmadfirozbin@gmail.com
Article Publishing History
Received: 22/10/2020
Accepted After Revision: 12/12/2020
Protein function depends on interaction with their ligands and mannose is one of the important ligands for understanding glycoprotein, so it is often required to calculate the binding site residues in protein at different distance threshold from PDB file. To study particular protein chain and its interaction with mannose in complex form, researchers have to parse the output of different available tools or databases for binding-site residues. Here we have developed a tool for calculating amino acid contact distances in proteins at different distance threshold using PDB file. ContMann can quickly find all binding-site residues in the protein by calculating distances from its coordinate present in pdb file by selecting the different distance threshold, Additionally, it can also generate atomic details of contacts including distances of binding-site residue. ContMann tool is available at: http://procarb.org/procarbdb/cfind-contact2.html
Protein Function Depends, Contacts Including Distances.
Alomrani A. A. S, Ali H. M, Firoz A. Contmann: A Tool to Calculate Contact Distances Between Amino Acid and Mannose Using Protein Data Bank File at Distance Cutoff. Biosc.Biotech.Res.Comm. 2020;13(4).
Alomrani A. A. S, Ali H. M, Firoz A. Contmann: A Tool to Calculate Contact Distances Between Amino Acid and Mannose Using Protein Data Bank File at Distance Cutoff. Biosc.Biotech.Res.Comm. 2020;13(4). Available from: <a href=”https://bit.ly/39IysL4″>https://bit.ly/39IysL4</a>
Copyright © Othman et al., This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) https://creativecommns.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
The proteins function depends on interaction with their ligands, among ligands Mannose is very important (Turner MW, 2003, Ng KK, 2002). Therefore, identifying amino acid contacts is important for understanding the glycoproteins. In order to understand the interactions calculating the amino acid contacts at different distance thresholds required ( Kenneth, 2002). Binding site residues of proteins can also be identified from databases (Desaphy 2015), visualization tools (Jendele 2019), or many other web servers developed earlier (Jendele 2019, Angles 2020), but this becomes overwhelmingly imposing when a large set of proteins have to be analysed. With the help of this tool, user can get the binding residue by after uploading PDB file. Additionally, it can also generate atomic details of contacts including distances of binding-site residue from PDB structures. Protein Data Bank (PDB) is repository of for 3D structures of biological macromolecules which has coordinates of its atoms (Kayikci 2018; Berman 2003), using these coordinates of two atoms, this tool can compute the distance between them.
A residue is defined as a binding residue if the distance between atoms of the interacting partner is less than a certain distance cut off (Eyal 2001). Upon uploading the protein mannose 3D-structure file of interest and option selected for distance threshold by user, ContMann searches the PDB file for the protein chains, Ligand chain of interest and the number of protein models (if multi model protein). If more than one model is present, ContMann gives option to select and parse the desired model present in the uploaded Protein Data Bank (PDB) file. Then ContMann calculates the distance between selected protein chain residue atoms and interacting partner atoms, and when this distance falls below or equal to the selected distance threshold, this residue is considered as binding residue. The overall description is illustrated at home page (Figure 1).
Web Interface: Web interface of current version of ContMann developed using HTML, JavaScript and CGI-PERL scripting language. It has home page where description about this tool mentioned, uploading pdb file option and submit button provided, here user can also select distance threshold in angstrom (Figure-1).
Figure 1: Screen shot of ContMann Home page. From this home page user can upload PDB file and select distance threshold.
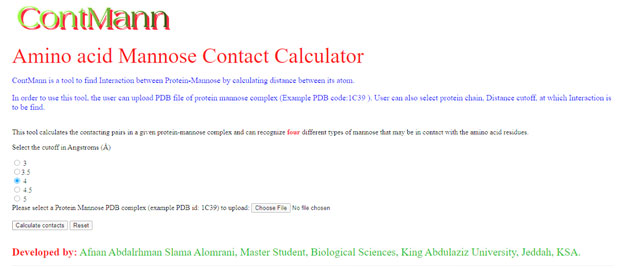
Program input: The input to the ContMann is a protein 3D coordinate PDB file, modeled protein or a docked complex file. The user can select provided distance threshold also (figure 1).
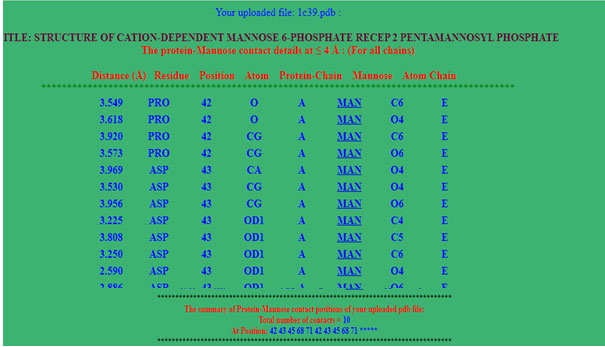
Program output: At the top of the program’s result page (figure 2), the uploaded file name and distance threshold selected for the calculations are displayed. The calculated distance between the two atoms, its residue, protein chain and the interacting atoms is displayed in a tabular form (figure2). The page summary section has total number of contacts at the end of page. First five column has distance, residue, position, atom and chain of protein and last 3 column has ligand three letter code, atom and chain of ligand. At the end of output summary of binding residue also mentioned.
Figure 2: Screen shot of output page. Here user can find the interaction residues with its atom, distances in angstrom and summary of Amino acid contact with mannose.
CONCLUSION:
The developed tool will be useful for the identification and analysis of binding sites residue of protein from 3D-structure PDB file of protein mannose complex at different distance threshold. Although, in current version only one pdb file can be uploaded but it will be upgraded for batch file, so user can upload list of files.
ACKNOWLEDGEMENTS
This work was not supported by any funding agency. We acknowledge with thanks Bioinformatics and Computational Biology Unit at Department of Biological Sciences, King Abdulaziz University, Jeddah for providing their support and providing facilities.
REFERENCES
Angles R, Arenas-Salinas M, García R. et al (2020). GSP4PDB: a web tool to visualize, search and explore protein-ligand structural patterns. BMC Bioinformatics 21, 85
Berman HM, Henrick K, Nakamura H (2003) Announcing the worldwide Protein Data Bank Nature Structural Biology 10 (12): 980.
Desaphy J, Bret G, Rognan D et al (2015). sc-PDB: a 3D-database of ligandable binding sites—10 years on, Nucleic Acids Research, Volume 43, Issue D1, D399–D404
Eyal E, Najmanovich R, Sobolev V, Edelman M (2001). MutaProt: a web interface for structural analysis of point mutations. Bioinformatics. 17(4):381-2.
Ezekowitz R A (2003), Role of the Mannose-Binding Lectin in Innate Immunity, The Journal of Infectious Diseases, Volume 187, 2003, S335–S339
Jendele L, Krivak R, Skoda P et al (2019). PrankWeb: a web server for ligand binding site prediction and visualization, Nucleic Acids Research, Volume 47, Issue W1, W345–W349
Karplus M and Kuriyan J (2005). Molecular dynamics and protein function. Proc. Natl. Acad. Sci. 102, 6679–85
Kayikci M, Venkatakrishnan AJ, Scott-Brown J, Ravarani CNJ, Flock T, Babu MM (2018). Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nat Struct Mol Biol. 25(2):185-194
Kenneth Ng, Kolatkar AR, Park-Snyder S, Feinberg H, Clark DA, Drickamer K, Weis WI (2002). Orientation of bound ligands in mannose-binding proteins. Implications for multivalent ligand recognition. J Biol Chem.277(18):16088-95.
Turner MW (2003). The role of mannose-binding lectin in health and disease. Mol Immunol. 40(7):423-9

