1Laboratory of Environment and Biotechnology, Department of Botany, Patna University, India
2Department of Biochemistry, Patna University, India
Corresponding author email: shardendu77@rediffmail.com
Article Publishing History
Received: 17/10/2020
Accepted After Revision: 18/12/2020
The treatment of wastewater technologies depends on a composition of physio- chemical as well as biological factors. Bacteria present in wastewater treatment plants, which may play an essential role for the degradation, mitigation and removal of organic waste and xenobiotic pollutants. Many of the bacterial species have been useful in complementary treatments for the domestic effluents which are rich in lipids and oils. Bacterial lipases have significant industrial, domestic etc. attention because of their stability, substrate specificity, high yielding capacity, and regular supply, as well as the fact that the microorganisms producing them and grow rapidly on inexpensive media. Bacterial biodiversity studies were done using 16S r RNA v3-v4 region of a domestic liquid waste collected from high density and average per capita income ($ 2000) population of eastern India (25°37’19.12’’ N, 85° 10’ 07.85” E). Next Generation Sequencing (NGS) data set revealed the total 180,933 filtered reads.
Analysis of v3-v4 region of filtered reads indicates that species are placed in 9 phyla, 15 classes, 20 orders, 20 families, 20 genus’s and 20 species. Among them most dominant phylum was firmicutes having 51% diversity and Chloroflexi (0.01%) had minimum domination. At the class level Bacillales (43%) was dominant, whereas at order and family level bacilli and streptococcus were dominant respectively. Lactococcus was most dominant species and surprisingly placed in unclassified group. It has been concluded that the niche of Janthinobacterium lividum and Acinetobacter jonshoni was isolated and identified in this tropical region. However, Janthinobacterium lividum shows the capnophilic behaviour, antitumor properties and its growth favoured by the enhancement of CO2 concentration. While Acinetobacter jonshoni causes human skin infection by colony formation and catheter related blood stream infection. So, we can recommend these bacterial strains to medical as well as in an industrial application for human health concern.
– Bacterial biodiversity, 16S r RNA, Janthinobacterium lividum, Bacillales, v3-v4, NGS.
Kumar N, Shardendu. Application of 16 Rrna Gene of V3-V4 Region for Meta Barcoding of Bacterial Community in High Density Population of Eastern India. Biosc.Biotech.Res.Comm. 2020;13(4).
Kumar N, Shardendu. Application of 16 Rrna Gene of V3-V4 Region for Meta Barcoding of Bacterial Community in High Density Population of Eastern India. Biosc.Biotech.Res.Comm. 2020;13(4). Available from: https://bit.ly/2GxF2rG
Copyright © Kumar and Shardendu This is an Open Access Article distributed under the Terms of the Creative Commons Attribution License (CC-BY) https://creativecommons.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
Bacterial biodiversity is an essential contributor which converts complex organic or toxic components in domestic liquid waste treatment system. As well as these are also essential for the optimal operation and conservation of Biological treatment systems (Moura, 2009). The biological community of activated sludge has a large biological diversity and contains a variety of viruses, bacteria, protozoa, fungi, algae, and metazoan. In this complex ecosystem, bacteria typically account for 95% of the total number of microbes and play a crucial part in wastewater treatment system. Nowadays studies of microbiome often give credence to the analysis of 16S ribosomal RNA sequences for the taxonomic identification of bacterial and archaeal strains. Approximately 1600 base pairs long are present on 16S rRNA gene which includes nine hypervariable regions of various conservation (V1-V9) (Handelsman, 2004; Ortiz- Alvarez et al., 2016). More conservative regions are useful for determining the higher-ranking taxa, whereas more quickly evolving ones can help identify genus or species. Metabarcoding using 16S rRNA marker is widespread in the studies of various microbial communities (Bolger et al., 2014; Babilonia et al., 2018).
The introduction of the next-generation sequencing techniques has led to novel applications of metabarcoding methods. In particular, increased read counts have allowed for quantitative estimates of the microbial community composition. Another advantage of NGS-based metabarcoding is that quantitative analysis has become available for communities of uncultured microbes. Yet, using NGS technologies has its limitations, caused chiefly by shorter read length. Its most important impact is the decreased precision of species identification. Domestic liquid wastes contain various type of bacteria and other micro-organisms originating from human wastes and other sources. Many of these bacteria are beneficial and are responsible for the biodegradation of organic components of the wastes whereas some others are may be pathogenic.
Normally bacterial species present in wastewater may cause pathogenicity which infect to the society through food and water. The safe management and disposal of any waste containing human excreta is the most critical aspect of sanitation and hygiene. The sample of domestic waste was collected from bank of river Ganga GPS (Global Positioning System) 25°37’19.12’’ N, 85° 10’ 07.85” E, the sample collection site is a heart of city, having population density of 1803 per square kilometre and per capita income is 671$ (Cardoso et al. 2017; Babilonia et al., 2018).
Domestic waste treatment system is available only to few percentages of human population. Domestic liquid wastes contain various type of bacteria and other micro-organisms originating from human wastes and other sources. Analysis of bacterial biodiversity in domestic liquid waste water, the variation in bacterial community in domestic liquid waste is temporally, while it mainly effects on human welfare as well as public health (Veldhuis et al., 2010; Taylor et al., 2011). The safe management and disposal of any waste containing human excreta is the most critical, aspect of sanitation and hygiene is essential to prevent the spread of infectious disease. Metagenomic studies gaining importance in providing a better understanding of microbial ecology and its applications in human welfare.
MATRIAL AND METHODS
We collected a domestic liquid waste (pH 8.4) aseptically in sterile glass bottle from house hold discharge near bank of river Ganga specifically identify the bacterial biodiversity. The GPS (Global Positioning System) location of both sample collection site was 25°37’19.12’’ N, 85° 10’ 07.85” E. Domestic waste water samples were collected and filtered through What-man filter paper No.1 with 0.7 μm retained the average particle size and it was stored at frozen temperature until the analysis.Metagenomic DNA was extracted from the Domestic liquid waste samples by commercially available Nucleospin Soil Kit (Eurofins Genomic lab, Bengaluru, India). The qualities of the isolated metagenomic DNA sample were quantified by using NanoDrop. Sanger sequencing data deposited in GeneBank under the accession numbers PRJNA557310 for bacteria.
Preparation of 2×300 MiSeq library (Illumina MiSeq Next Generation Sequencer) was done using the amplicon libraries were prepared using Nextera XT Index Kit (Illumina inc.) as per the 16S Metagenomic Sequencing Library Preparation protocol (Part # 15044223 Rev.B). Primers for the amplification of the bacterial 16S V3-V4 region were designed and synthesized at Eurofins Genomics Lab. There was approximately 300 base- pair reads generated from each forward and reverse direction which results that entire targeted region having nearly double coverage. Sequence reads were assembled which generates Operational Taxonomic Unit (OTU) that was based on 97% sequence similarity, and two different set of DNAs (chimera) were removed by v 0.38 (Edgar, 2010; Edgar et al., 2011).
Amplification of the 16S gene was carried out. 3μl of PCR product was resolved on 1.2% Agarose gel at 120V for almost 60 minute or till the samples reached 3/4th of the gel. Primers used in present study was 16S rRNA F-GCCTACGGGNGGCWGCAG and 16S rRNA R- ACTACHVGGGTATCTAATCC. The quality control (QC) passed amplicon with the Illumina adaptor was amplified using i5 and i7 primers that added multiplex index sequence as well as common adapters required for cluster generation (P5 and P7) as per the standard Illumina protocol. The amplicon libraries were purified by AMPureXP beads technique and quantified by using Qubit fluorometer. Identified DNA sequences for every OTU were align by using QIIME (Caporaso et al., 2010). Taxonomic distribution of each respective sequence was assigned on the basis of Ribosomal Database Project (RDP) taxonomy (http://rdp.cme.msu.edu).
Quantity and quality check (QC) of library on Agilent 4200 Tape Station was done using only those samples which had mean peak value of amplification of genes obtained through Tape screening method were further used for cluster generation sequencing. The Agilent 4200 Tape Station system was an established automated electrophoresis tool for DNA and RNA sample quality control. Fully automated sample processing enables the unattended analysis of size, concentration and integrity. The Tape Station system provides a complete solution for true end-to-end sample quality control within any next-generation sequencing (NGS). This covers the full DNA and RNA range and offers high levels of flexibility in a simplified workflow with ready-to-use Screen Tapes. The amplified libraries were analysed on 4200 Tape Station system (Agilent Technologies) using D1000 Screen tape as per manufacturer instructions. D1000 screen tape assay is referred to analysing DNA molecules from 35-1000bp (Ortiz-Alvarez and Casamayor 2016).
Cluster Generation and Sequencing was done after obtaining the mean peak size from Tape Station profile, the libraries were loaded onto MiSeq at appropriate concentration (10-20pM) for cluster generation and sequencing. The MiSeq is an integrated instrument which amplify the clone, sequencing of genomic DNA as well as data analysis with alignment, variant calling, base calling in a single run. Paired-End sequencing allows the template fragments to be sequenced in both the forward and reverse directions on MiSeq. The kit reagents were used in binding of samples to complementary adapter oligo on paired-end flow cell. They were designed to allow selective cleavage of the forward strands after re-synthesis of the reverse strand during sequencing.
The copied reverse strand was then used to sequence from the opposite end of the fragment. The data generated during our study have been deposited in the NCBI (National Centre for Biotechnology Information) Sequence Read Archive (SRA) that are available under the Bio-Sample accession numbers SAMN12393872 and SAMN12393873. The 16S rRNA gene amplicon data generated during our study have been deposited in the NCBI (National Centre for Biotechnology Information) Sequence Read Archive that are available under the Bio-Sample accession numbers SAMN12393872 (Ortiz-Alvarez and Casamayor 2016).
Bioinformatic analysis was done through QIIME (canonically pronounced ‘chime’). It is software that performs microbial community analysis. It is an acronym for Quantitative Insights into Microbial Ecology (QIIME), and has been used to analyse and interpret nucleic acid sequence data from fungal, viral, bacterial, and archaeal communities. The QIIME is comprehensive software comprising of tools and algorithms such as FastTree for heuristic based maximum likelihood phylogeny inference (Price Morgan N., et. al., 2010), the Ribosomal Database Project (RDP) classifier for the assignment of taxonomic data using a naïve Bayesian classifier (Wang et al., 2007) and others. This allows QIIME, which continues to undergo development, to easily and relatively adapt standalone tools, and thus improve in step with advances in the field of microbial community ecology (Fig.2) (Ortiz-Alvarez and Casamayor 2016).
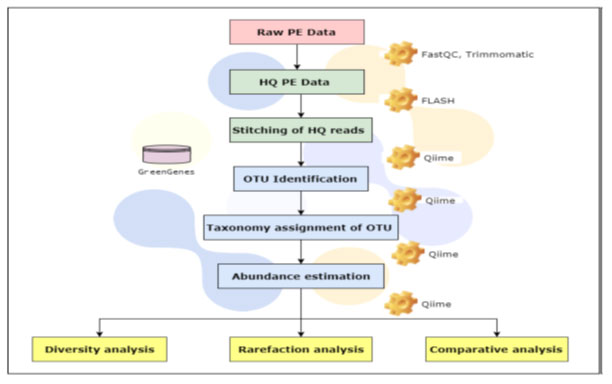
RESULTS AND DISCUSSION
The total genomic DNA was isolated from the water-soil mixture using a Nucleospin Soil Kit (Eurofins Genomic lab, Bengaluru, India). The qualities of the isolated metagenomic DNA sample were quantified by using Nanodrop in Table-1.
Table 1. Quantification of Metagenomic DNA in domestic liquid waste
| Sample | Nano Drop Reading(ng/μl) | Nano Drop (O.D.A260/280) | Nano Drop (O.D. A260/230) |
| Domestic liquid waste | 127.9 | 1.82 | 1.65 |
The amplicon library was prepared using a Next era XT index kit (Illumina, Inc.). The primers for the amplification of the V3-V4 region of 16S rRNA genes (forward, GCCTACGGGNGGCWGCAG, and reverse, ACTACHVGGGTATCTAATCC) were designed at Eurofins. The V3-V4 region is highly variable and known to reveal microbially diverse populations. The amplicon library was purified using 1 AM Pure XP beads, quantified using a Qubit fluorometer, and analysed with a 4200 Tape Station system (Agilent Technologies) using D1000 screen tape following the manufacturer’s instructions. The library was loaded onto a MiSeq instrument (2 x 300 bp) at 10 to 20 pM for cluster generation and sequencing. The sequenced raw reads were processed to obtain high-quality (HQ) reads using Trimmomatic v0.35 (5) to remove adaptor sequences, ambiguous reads, and low-quality sequences (i.e., those with a more than 5% quality threshold [QV] of <20 Phred quality score). A total of 180,933 high-quality reads were obtained (Yankson and Steck 2009; Ortiz-Alvarez and Casamayor 2016).
The HQ reads were subjected to operational taxonomic unit (OUT) identification at 97% sequence similarity and taxonomic assignment of OTUs using the Green genes database (16S/Archaea database) and the Quantitative Insights into Microbial Ecology (QIIME) module. Krona-based (6) diagram visualization showed that 99.22% of the microbially diverse population represents. When sample of domestic liquid waste were put to Next Generation Sequencing (NGS), large number of bacteria were identified. All of them were grouped into 9 phyla. Out of 9 phyla, Firmicutes, Proteobacteria and Bacteroidetes were most abundant phylum in domestic liquid waste (Ortiz-Alvarez and Casamayor 2016).
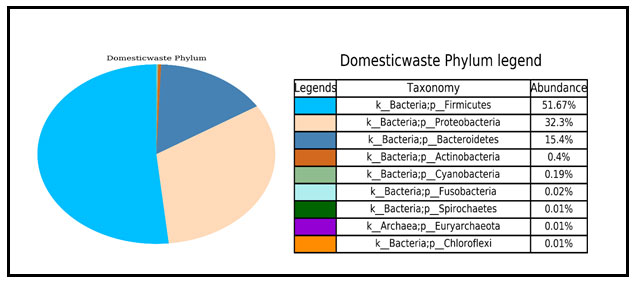
Phylum level: At the phylum level bacterial were constitutes Firmicutes phylum (51.67%), Proteobacteria phylum (32.3%) and Bacteroidetes phylum (15.4%) in domestic waste sample (Fig 1). Betaproteobacteria was constituents of Proteobacteria responsible for degradation of organic matter and nutrient cycling.
Figure 1: Pie chart showing the absolute abundance of the sample at phylum level within each bacterial community. It is shown that the most abundant phylum is firmicutes.
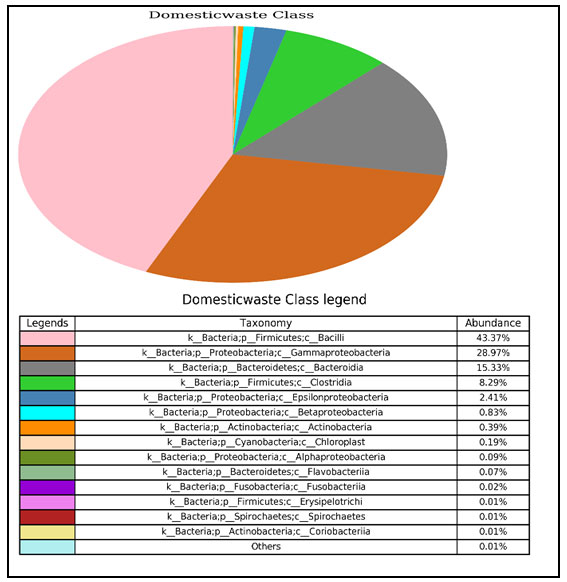
Identification of bacteria at class level in domestic liquid waste sample through NGS: Now sample were analysed for class level. In which identified bacteria were belongs to 15 classes irrespective of their percentages at phylum level. Out of 15 classes, four classes were dominated and their percentage of occurrence of bacteria as follow Bacilli (43.37%), Gamma proteobacteria (28.97%), Bacteroidia (15.33%) and Clostridia (8.29%) (Fig 2).
Figure 2: Pie chart shows their abundance in percentage of each class within each bacterial community.
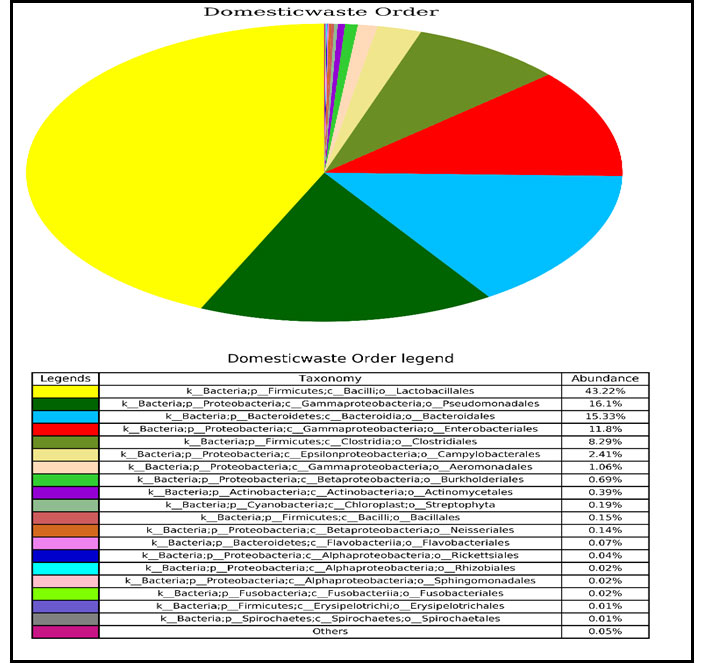
Identification of bacteria at order level in domestic liquid waste sample through NGS: It has been found that Lactobacillales was most abundant order of bacteria within each microbial community. The percentage of abundance of bacteria at order level such as Lactobacillales (43.22%), Pseudomonadales (16.1%), Bacteriodales (15.33%), Enterobacterials (11.8%) and Clostridiales (8.29%) were found in domestic liquid waste (Fig.3).
Figure 3: Pie chart showing the absolute abundance of each order within each bacterial community
Identification of bacteria at family level in domestic liquid waste sample through NGS: It has been found that Streptococcaceae was most abundant family within each microbial community in taxonomic distribution of domestic waste. The percentage of abundance of bacteria at family level were Streptococcaceae (30.01%), Pseudomonadaceae (15.76%), Carnobacteriaceae (12.94%), Enterobacteriaceae (11.8%), Porphyromonadaceae (10.36%) and Veillonellaceae (8.11%) (Fig.4).
Figure 4: Pie chart showing the absolute abundance of each family within each bacterial community.

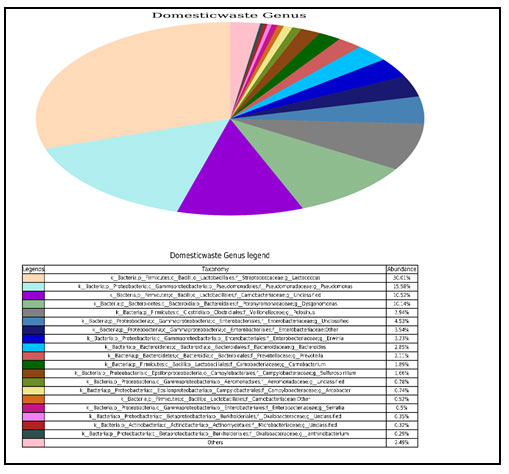
Identification of bacteria at genus level in domestic liquid waste sample through NGS: It can be inferred that the most dominated genus was Lactococcus in domestic waste. The absolute abundance of each genus within each microbial community were Lactococcus (30.01%), Pseudomonas (15.58%), Unclassified (10.52%), Dysgonomonas (10.14%), Pelosinus (7.94%) (Fig.5) (Ortiz-Alvarez and Casamayor 2016).
Figure 5: Pie chart showing the absolute abundance of each genus within each bacterial community. From the figure, it can be inferred that the most abundant genus is Lactococcus.
Identification of bacteria at species level in domestic liquid waste sample through NGS: It has been found that the most abundant bacteria at species level taxonomic distribution was unclassified bacteria but at genus level it belongs from Lactococcus. The percentage of abundance within each bacterial community were Unclassified from Lactococcus genus (30.01%), Unclassified from Pseudomonas genus (15.29%), Unclassified from Carnobacteriaceae family (10.52%), Unclassified from Dysgonomonas genus (10.14%) (Fig.6) (Yankson and Steck 2009).
Figure 6: Pie chart showing the absolute abundance of each species within each bacterial community.
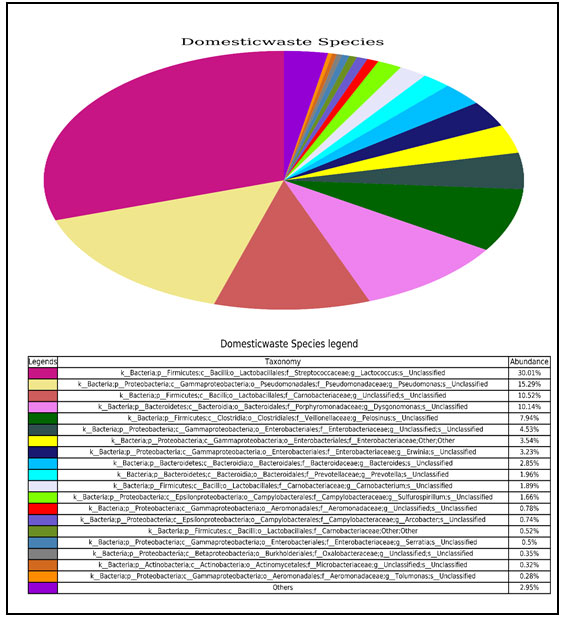
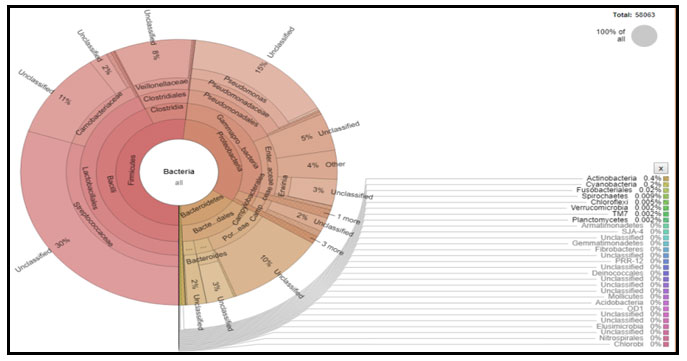
Figure 7: Krona chart of the bacteria represented by 16S rRNA gene amplicon-based bacterial diversity in domestic liquid waste. Each circle represents the phylum, class, order, family, genus, and species from the inside to the outside of the circle, respectively, indicated by percent diversity based on the absolute number of representative bacteria.
It can be inferred that the most dominated genus was Lactococcus in domestic waste. The absolute abundance of each genus within each microbial community were Lactococcus (30.01%), Pseudomonas (15.58%), Unclassified (10.52%), Dysgonomonas (10.14%), Pelosinus (7.94%). The data representing Firmicutes (0.05088%), Proteobacteria (0.2445%), and Bacteroidetes (0.148%), was classified to the species level (Table 2) (Yankson and Steck 2009).
Table 2. Summary of bacteria identified to the species level from 16S RNA gene- based metagenomic study of a domestic liquid waste collected from near to PMCH, Patna Bihar, India
| Phylum | Class | Order | Family | Genus | Species | Absolute Count | % |
| Proteobacteria | Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Acinetobacter | Johnsonii | 142 | 0.2445 |
| Betaproteobacteria | Burkholderiales | Oxalobacteraceae | Janthinobacterium | Lividum | 138 | 0.2377 | |
| Gammaproteobacteria | Pseudomonadales | Pseudomonadaceae | Pseudomonas | Viridiflava | 76 | 0.1309 | |
| Gammaproteobacteria | Pseudomonadales | Pseudomonadaceae | Pseudomonas | Fragi | 70 | 0.1206 | |
| Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Acinetobacter | rhizosphaerae | 5 | 0.0086 | |
| Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Psychrobacter | Sanguinis | 3 | 0.0052 | |
| Alphaproteobacteria | Rhizobiales | Methylobacteriaceae | Methylobacterium | Adhaesivum | 3 | 0.0052 | |
| Bacteroidetes | Bacteroidia | Bacteroidales | Prevotellaceae | Prevotella | Copri | 83 | 0.1429 |
| Bacteroidia | Bacteroidales | Prevotellaceae | Prevotella | Stercorea | 2 | 0.0034 | |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Faecalibacterium | Prausnitzii | 3 | 0.0052 |
| Clostridia | Clostridiales | Veillonellaceae | Veillonella | Dispar | 1 | 0.0017 | |
| Actinobacteria | Coriobacteriia | Coriobacteriales | Coriobacteriaceae | Collinsella | Aerofaciens | 3 | 0.0052 |
| Actinobacteria | Actinomycetales | Micrococcaceae | Rothia | mucilaginosa | 1 | 0.0017 | |
| Actinobacteria | Actinomycetales | Corynebacteriaceae | Corynebacterium | Variabile | 3 | 0.0052 | |
| Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Verrucomicrobiaceae | Akkermansia | Muciniphila | 1 | 0.0017 |
| Fusobacteria | Fusobacteriia | Fusobacteriales | Fusobacteriaceae | Cetobacterium | Somerae | 2 | 0.0034 |
Our study site revealed that the partially treated domestic liquid waste, which also creates a carbon source of microbes and results is huge bacterial diversity. In our knowledge, very little or no explicit work on flood plain bacterial biodiversity by Next Generation Sequencing (NGS) is available. So, we attempted this study to reveal bacterial biodiversity in such a complex ecosystem/niches by NGS. The software of Illumina-based, as per the 16S Metagenomic Sequencing Library preparation protocol, near full-length and differential diversity studies.
Metagenomic DNA was isolated from the collected sample of domestic liquid waste (DLW). Extraction of DNA is a vital step because it can affect on both the quality and quantity of DNA extracted. Furthermore, an increase the concentration of DNA yield is important to ensure that the DNA sample is representative of the gene pool (A gene pool is the collection of total genes within an interbreeding population) of the collected sample. The extraction of DNA yield may be depending on the physical properties and chemical composition of the matrix. The concentration of quantified DNA was found in domestic liquid waste as 127.9 (ng/μl) by Nano drop readings which may indicate the more bacterial diversity in their community (Zhou et al., 1996; Lakay et al., 2007; Yankson and Steck 2009).
Inspite of that the amount of extracted DNA purity of examined by the ratios A260/280 and A260/230 and the integrity of the extracts checked by agarose gel electrophoresis, are also important parameters for gene amplification by PCR (von Wintzingerode et al., 1997). When the ratio of A260/230 is low that indicates the protein contamination in DNA extraction from the samples which reflected the co-extraction of contaminants absorbing at 230 nm such as residual phenol from nucleic acid extraction and humic acid and fulvic acids (Whitehouse and Hottel 2007; Techer et al., 2010). So, in our sample we got quantification of isolated metagenomic DNA sample on nanodrop was the ratio of A260/230 -1.65 in domestic liquid waste.
The sequenced library was further analyzed through bioinformatics tools. There were used a software for the data analysis called as QIIME software. QIIME (canonically pronounced ‘chime’) is software that performs microbial community analysis. It is an acronym for Quantitative Insights into Microbial Ecology (QIIME), and has been used to analyses and it also interpret nucleic acid sequence data from fungal, viral, bacterial, and archaeal communities. Operational Taxonomic Unit (OTUs) are clusters of sequences, frequently intended as 97% sequence similarity. Bioinformatics studies and taxonomic profiling revealed existence complex of bacterial population at the phylum level comprising of Frmicutes, Proteobacteria, Bacteriodetes, Actinobacteria, Chloroflexi etc. in domestic liquid waste.
From the analysis, it was conferred that the phylum Firmicutes (51.67%), was most abundant in domestic liquid Waste. samples exhibited high degree of diversity at class level. The prominent communities identified were Bacilli, Gamma proteobacteria, Bacteroidia, Clostridia, Alpha proteobacteria, Delta proteobacteria, Thermoleophilia, Planctomycetia etc. Bacilli (43.37%). Study also revealed presence of great diversity at the order level such as Lactobacillales, Pseudomonadales, Bacteriodales, Enterobacteriales, Clostridiales, Actinomycetales, Bacillales, Rhizobiales etc. in domestic liquid waste, Out of these identified bacteria at class level, Lactobacillales (43.22%). We have found that Streptococcaceae (30.01%) was relatively most abundant bacteria at phylum level of taxonomic distribution in DLW.
Unclassified bacteria at the genus level were found in both samples. Unclassified belongs to genus belongs to Carnobacteriaceae (10.52%), Nocardioidaceae (17.53%) and Micrococcaceae (5.62%) family. After that Alpha diversity summarizes the diversity of organisms in a sample with a single number. This diversity of annotated samples can be estimated from the distribution of the species – level annotations. Shannon alpha diversity of domestic waste was 4.822. Our study revealed previously unknown groups of bacteria highlighting the continued importance of general culture independent 16S rDNA surveys of unexplored environments. These novel bacteria make up a substantial proportion of the diversity sampled and are likely to play key roles in Himalayan soil biogeochemistry.
CONCLUSION
This study provides essential baseline information on bacterial communities in the domestic liquid waste (near PMCH, Bihar). The most abundant identified bacterial biodiversity from phylum to species level in the domestic liquid waste. The relative abundance of bacterial diversity differed significantly according to the DNA extraction method used; so, the results of studies using different methods need to interpretation. This result will be the selection of an appropriate DNA extraction for metagenomics studies which help to st6 different toxic as well as.
ACKNOWLEDGMENTS
We acknowledge the Laboratory of Environment and Biotechnology, Patna University, and Ministry of Environment, Forest and Climate Change, Bihar for supporting this work.
Conflict of Interest: The author has no conflict of interest.
REFERENCES
Babilonia J, Conesa A, Casaburi G, Pereira C, Louyakis AS, Reid RP, and Foster JS. (2018). Comparative metagenomics provides insight into the ecosystem functioning of the Shark Bay stromatolites, Western Australia. Front Microbiol 9:1359. https://doi.org/10.3389/fmicb.2018.01359.
Bolger AM, Lohse M, and Usa del B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170.
Caporaso, J.G., Kuczynski,J., Stombaugh, J., Bittinger, K., Bushman, F.D. and Knight, R. (2010) QIIME allows analysis of high throughput community sequencing data. Nat. Methods 7, 335-336.
Cardoso DC, Sandionigi A, Cretoiu MS, Casiraghi M, Stal L, and Bolhuis H. (2017). Comparison of the active and resident community of a coastal microbial mat. Sci Rep 7:2969. https://doi.org/10.1038/s41598-017-03095-z.
Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics.; 26(19):2460–1. pmid:20709691.
Edgar RC, Haas BJ, Clemente JC, Quince C, and Knight R (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics.;27(16):2194–200. pmid:21700674.
Handelsman J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68:669–685. https://doi.org/10.1128/MMBR.68.4.669-685.2004.
Lakay FM, Botha A., and Prior BA (2007). Comparative analysis of environmental DNA extraction and purification methods from different humic acid-rich soils. J. Appl. Microbiol.102: 265 273.
Moura, A., Tacão, M., Henriques, I., Dias, J., Ferreira, P., and Correia, A., (2009). Characterization of bacterial diversity in two aerated lagoons of a waste water treatment plant using PCR-DGGE analysis. Microbiol. Res. 164, 560–569, http://dx.doi.org/10.1016/ j. micres. 2007.06.005.
Ondov BD, Bergman NH, and Phillippy AM. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. https://doi .org/10.1186/1471-2105-12-385.
Ortiz-Alvarez R, and Casamayor EO. (2016). High occurrence of Pacearchaeota and Woesearchaeota (Archaea super phylum DPANN) in the surface waters of oligotrophic high-altitude lakes. Environ Microbiol Rep 8:210 –217.https://doi.org/10.1111/1758-2229.12370.
Morgan N. P., Paramvir S. Dehal, A. P., and Arkin (2010), Fast Tree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. https://doi.org/10.1371/ journal. Pone .0009490.
Taylor J, Lai KM, Davies M, Clifton D, Ridley I, and Biddulph P (2011). Flood management: prediction of microbial contamination in large-scale floods in urban environments. Environment International.; 37 (5):1019–29. Epub 2011/04/13. doi: 10.1016/j.envint.2011.03.015 PMID: 21481472.
Techer D, Martinez-Chois C, D’Innocenzo M, Laval-Gilly P, Bennasroune A, and Foucaud L, (2010). Novel perspectives to purify genomic DNA from high humic acid content and contaminated soils. Sep. Purif. Technol. ;75: 81–86.
Ten Veldhuis JA, Clemens FH, and Sterk G (2010), Berends BR. Microbial risks associated with exposure to pathogens in contaminated urban flood water. Water research.; 44 (9):2910–8. Epub 2010/03/17. doi: 10.1016/j.watres.2010.02.009 PMID: 20227742.
Tringe, S. G. and Hugenholtz, P. A (2008) Renaissance for the pioneering 16S rRNA gene. Current Opinion in Microbiology 11, 442–446.
Von Wintzingerode F, Gobel UB., and Stackebrandt E (1997). Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev.;21: 213–229.
Wang Y, (2007) Human 1A6/DRIM, the homolog of yeast 20, functions in the 18S rRNA processing. Biochim Biophys Acta 1773 (6):863-8.
Wang, Y. and Qian, P. Y. (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PloS ONE 4, e7401.
Whitehouse CA., and Hottel HE (2007). Comparison of five commercial DNA extraction kits for the recovery of Francisella tularensis DNA from spiked soil samples. Mol. Cell. Probes. 21: 92–96.
Yankson KK., and Steck TR (2009). Strategy for extracting DNA from clay soil and detecting a specific target sequence via selective enrichment and real-time (quantitative) PCR amplification. Appl. Environ. Microbiol.; 75: 6017–6021.
Zhou JZ, Bruns MA., and Tiedje JM (1996). DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. ;62: 316–322.

