Renu1, Sirajuddin Ali1, Ajaj Hussain1, Sukrit Srivastava2, Mohit Kamthania*1![]() and Abhimanyu Kumar Jha1
and Abhimanyu Kumar Jha1
1Department of Biotechnology, Institute of Applied Medicines and Research, Ghaziabad, India
2Infection Biology Group, Indian Foundation for Fundamental Research, RaeBareli, India
Corresponding author email: kamthania.mohit@gmail.com
Article Publishing History
Received: 23/10/2020
Accepted After Revision: 12/12/2020
H9N2 virus outbreak has increased worldwide in last decade due to the zoonotic potential of these viruses. H9N2 virus causes low pathogenicity but when co-infected with other pathogen it causes high mortality. Since 1998, H9N2 infection has Caused one death and more than 59 cases as reported worldwide in animals including humans. There are currently no clear methods to control the pandemic potential of H9N2 virus globally, so there is urgent need for vaccine designing against these viruses.In this study, screening of T-Cell epitopes from H9N2 virus proteins viz nuclear export protein, nonstructural protein 1, matrix protein 2, matrix protein 1, neuraminidase, nucleocapsid, hemagglutinin, polymerase PA, PB1-F2 protein & polymerase PB2 protein followed by highest binding affinity of selected T-cell epitopes with their corresponding HLA alleles has been done. The server ProPred1 & ProPred facilitates the binding prediction of HLA class I & class II allele with specific epitopes from the antigenic protein sequences of H9N2 virus. PEPstrMOD server was used structure modeling of the screened epitopes. We docked the selected T-cell epitopes with their corresponding HLA allele structures using the HPEPDOCK Server. Toxicity & immunogenicity of epitopes were analyzed by Toxin Pred and IEDB tools, respectively. The screened T-cell epitopes viz FQGRGVFEL, AEIEDLIFL, IIEGRDRTL, RRVDINPGH, YIGVKSLKL, LVMKRKRDS, VVLVMKRKR, LVRKTRFLP are anticipated to be valuable in designing comprehensive epitope-based vaccines against H9N2 virus after further in-vivo studies.
H9N2 Virus, T-Cell Epitope, HLA Alleles, Vaccine Designing.
Renu, Ali s, Hussain A, Srivastava S, Kamthania M, Jha A. K. In-silico Sreening of T-Cell Epitopes as Vaccine Candidate from Proteome of H9N2 Virus. Biosc.Biotech.Res.Comm. 2020;13(4).
Renu, Ali s, Hussain A, Srivastava S, Kamthania M, Jha A. K. In-silico Sreening of T-Cell Epitopes as Vaccine Candidate from Proteome of H9N2 Virus. Biosc.Biotech.Res.Comm. 2020;13(4). Available from: <a href=”https://bit.ly/3gWYtHX”>https://bit.ly/3gWYtHX</a>
Copyright © Renu et al., This is an Open Access Article distributed under the Terms of the Creative Commons Attribution License (CC-BY) https://creativecommons.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
H9N2 viruses cause worldwide infections and the majority of confirmed cases are young children. Different combination of hemagglutinin and neuraminidase, surface proteins of Influenza A viruses, give rise to subtypes viz H1N1, H5N6, or H9N2. Different studies have showed that the primary routes of transmission of H9N2 virus are respiratory and direct contact. Aerosol, droplet particles, oral-facial route and direct touch are the other routes of transmission for this virus (Killingley et al., 2013). H9N2 virus infection has been observed in humans in Hong Kong, India, Bangladesh, Pakistan, Oman, Egypt and China (Butt et al., 2003; Shanmuganatham et al., 2013; Pan et al., 2018; Ali et al., 2019; Potdar et al., 2019). H9N2 outbreaks in commercial chickens from Asia, Middle East and African countries have also been reported recently (Li et al., 2020).
A recent report of H9N2 virus (strain A/India/TCM2581/2019) infection was observed in a 17 month-old boy residing at Melghat District, Maharashtra, India , which showed a threat to human infection in India and there is an urgent need for the treatment of this emerging virus (Potdar et al., 2019). Important implication of this study is to screen promiscuous T-cell epitopes from H9N2 virus proteins viz nuclear export protein, nonstructural protein 1, matrix protein 2, matrix protein 1, neuraminidase, nucleocapsid, hemagglutinin, polymerase PA, PB1-F2 protein & polymerase PB2 protein. The screened selected T-cell epitopes may be the promising targets for epitope-based vaccine design for H9N2 virus.
MATERIAL AND METHODS
Complete genome sequence of H9N2 virus strain (A/India/TCM2581/2019/(H9N2) was taken in this work (Potdar et al., 2019). The amino acid sequence of nuclear export protein, nonstructural protein 1, matrix protein 2, matrix protein 1, neuraminidase, nucleocapsid, hemagglutinin, polymerase PA, PB1-F2 protein and polymerase PB2 protein of H9N2 virus were retrieved from protein sequence database from NCBI (http://www.ncbi.nlm.nih.gov/protein) and their accession number were shown in Table 4. A proteomics server, ExPASy ProtParam (www.expasy.org) was used to analyze the primary structure of the target protein. Several parameters given by ProtParam tool for example estimated half-life, amino acid composition, theoretical pI, molecular weight, extinction coefficient, atomic composition, aliphatic index, grand average of hydropathicity (GRAVY) and instability index were examined. SOPMA (Geourjon and Deleage, 1995) server used to check the secondary structure (alpha helix, beta plated sheets, turns and coils) of the proteins, its aim to predict solvent accessibility, transmembrane helices, coiled-coil regions, globular regions and ultimately determines the stability and function of proteins.
To predict the protective antigens as vaccines, the sequence was then analyzed by VaxiJen.VaxiJen server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (Doytchinova and Flower, 2007) with default parameters to find out the antigenicity. All the antigenic proteins with their respective predicted score were computed. The prediction of potential HLA class I & class II binding nanomer epitopes completed by using Propred I & Propred respectively (Singh and Raghava, 2001). Threshold percentage of highest scoring peptides is taken at 3%. Top four binders for different HLA allele have been taken into consideration. Immunoproteosome site and Proteasome site filters were put in ‘on’ mode with threshold of 4% for each filter. ToxinPred (http://crdd.osdd.net/raghava/toxinpred/) was used to predict toxicity of predicted T- cell epitopes (Gupta et al., 2013).
ToxinPred is an in-silico tool to predict the selected epitope as toxic or non-toxic. ToxinPred was run with default parameters and only non-toxic T-cell epitopes were selected for further study. The PEPstrMOD (Singh et al., 2015) method performed to find out the tertiary structure of selected nanomer epitopes. The PEPstrMOD tool prediction strategy utilizes the secondary structure data & β-turns data anticipated by PSIPRED and BetaTurns respectively. The amino acid sequences of HLA alleles were retrieved from IMGT/HLA database (http://www.ebi.ac.uk/ipd/imgt/hla/intro.html) (Robinson et al., 2012) and homology model of alleles was constructed using program HPEPDOCK (http://huanglab.phys.hust.edu.cn/hpepdock/) Server (Zhou et al., 2018).
HPEPDOCK Server has been used to perform docking of epitopes with alleles models. Docking studies was performed to study the interaction of epitopes with alleles. For such interaction studies, the most important requirement was the proper orientation and conformation of epitope, which fit to the binding site of the allele appropriately and form the epitope-allele complex. The obtained docking scores was tabulated and analysed. Immunogenicity of the selected T-cell epitopes was predicted by using IEDB (Immune Epitope Database and Analysis Resource) (http://tools.iedb.org/immunogenicity/) (Calis et al., 2013). This tool predicts the relative ability of an epitope-HLA complex to elicit an immune response. Amino acid properties & their position within the epitope are utilized by this tool to predict the immunogenicity of a class I epitope-HLA complex.
RESULTS AND DISCUSSION
H9N2 viruses are emerging zoonotic infectious viruses that cause fatal diseases in both animals and humans (Pusch and Suarez, 2018). New efficient vaccines against H9N2 virus infection are urgently needed to control the disease and its proliferation. In the present study, prediction and modeling of T cell epitopes of H9N2 virus antigenic proteins followed by docking studies of predicted highest binding scores with their corresponding HLA class I and class II alleles have been performed.
Primary and secondary structure analysis: Primary structure analysis viz theoretical isoelectric point (PI), molecular weight, total number of positively charged residues (Arg+Lys) and negatively charged residues (Asp+Glu), estimated half-life (in vitro) in mammalian reticulocytes and instability index (II) are shown in table 1 while secondary structure analysis viz alpha helix, extended strand, beta turn & random coil are shown in table 2.
Table 1. Primary structure analysis using ProtParam
| Name of Protein | No. of amino acids | Molecular weight | Theoretical PI | Total no. of negatively charged residues(asp Glu) | Total no. of positively charged residues(as plys) | Extinction coefficient
|
Estimated half-life | Instability index | Aliphatic index | Grand average of hydropathi city |
| Nonstruc
tural protein 1
|
237 | 26966.89 | 5.51 | 36 | 31 | 34615 | 30 | 58.37 | 86.41 | -0.354 |
| matrix protein 2 | 97 | 11268.83 | 4.92 | 16 | 11 | 15595 | 30 | 57.86 | 92.37 | -0.242 |
| matrix
protein 1 |
252 | 27763.07 | 9.28 | 24 | 30 | 13075 | 30 | 36.32 | 85.99 | -0.218 |
| Neuramini
Dase |
469 | 51417.82 | 6.02 | 47 | 42 | 93610 | 30 | 33.69 | 75.22 | -0.280 |
| Nucleo
capsid |
498 | 56137.54 | 9.47 | 58 | 70 | 52745 | 30 | 44.25 | 70.76 | -0.561 |
| Hemagg
Lutinin |
560 | 62616.94 | 6.91 | 56 | 55 | 87165 | 30 | 31.37 | 84.61 | -0.345 |
| polymerase PA | 716 | 82568.39 | 5.54 | 112 | 95 | 95310 | 30 | 48.41 | 77.37 | -0.469 |
| polymerase PB2 | 759 | 85732.98 | 9.45 | 85 | 103 | 79090 | 30 | 45.89 | 87.80 | -0.298 |
Table 2. The secondary structure analysis using SOPMA
| Protein | Alpha helix | Extended strand | Beta turn | Random coil |
| Nonstructural protein 1 | 132(55.70%) | 30(12.66%) | 7(2.95%) | 68(28.69%) |
| matrix protein 2 | 49(50.52%) | 14(14.43%) | 4(4.12%) | 30(30.93%) |
| matrix protein 1 | 153(60.71%) | 24(9.52%) | 16(6.35%) | 59(23.41%) |
| Neuraminidase | 32(6.82%) | 162(34.54%) | 29(6.18%) | 246(52.45%) |
| Nucleocapsid protein | 218(43.78%) | 60(12.05%) | 31(6.22%) | 189(37.95%) |
| Hemagglutinin | 192(34.29%) | 117(20.89%) | 42(7.50%) | 209(37.32%) |
| polymerase PA | 388(54.19%) | 79(11.03%) | 30(4.19%) | 219(30.59%) |
| polymerase PB2 | 289(38.08%) | 147(19.37%) | 44(5.80%) | 279(36.76%) |
Protein antigenicity determination: Amino acid sequences of proteins viz nuclear export protein, nonstructural protein 1, matrix protein 2, matrix protein 1, neuraminidase, nucleocapsid, hemagglutinin, polymerase PA, PB1-F2 protein & polymerase PB2 were screened by VaxiJen. All the proteins were found antigenic except nuclear export protein & PB1-F2 protein which were non-antigenic at threshold value of 0.4 (default threshold for viral proteins) (Table 3). Antigenic proteins selected for further analysis.
Table 3. VaxiJen result of antigenicity
| S.No. | Protein | Overall Antigen Prediction |
| 1 | nuclear export protein | 0.3441 ( Probable NON-ANTIGEN ) |
| 2 | nonstructural protein 1 | 0.4290 ( Probable ANTIGEN ) |
| 3 | matrix protein 2 | 0.5641 (Probable ANTIGEN). |
| 4 | matrix protein 1 | 0.4805 ( Probable ANTIGEN ) |
| 5 | Neuraminidase | 0.5513 ( Probable ANTIGEN ) |
| 6 | nucleocapsid protein | 0.5208 ( Probable ANTIGEN ) |
| 7 | Hemagglutinin | 0.4322 ( Probable ANTIGEN ) |
| 8 | polymerase PA | 0.5273 ( Probable ANTIGEN ) |
| 9 | PB1-F2 protein | 0.1654 ( Probable NON-ANTIGEN ) |
| 10 | polymerase PB2 | 0.5291 ( Probable ANTIGEN ). |
Prediction and analysis of HLA Class I & Class II binding peptides: H9N2 virus proteins were subjected to Propred1 & Propred for selection of HLA Class I & HLA Class II specific T- cell epitopes binders respectively. Epitopes showing highest score with the maximum number of HLA alleles binders were selected at a threshold value of 3% (Table 4).
Table 4. ProPred1 & ProPred predicted T-cell epitopes for HLA Class I & Class II with binding scores
| Protein name | Amino acid length | Accession no. | Position | Epitopes | HLA class alleles | Propred (% of highest score) |
| nucleocapsid protein | 498 | QBP33428.1 | 457-465 | FQGRGVFEL | HLA-B*0705
|
3000.000
|
| nucleocapsid protein | 498 | QBP33428.1 | 250-258 | AEIEDLIFL | HLA-B*2705
|
3000.000
|
| polymerase PA | 716 | QBP33426.1 | 77-85 | IIEGRDRTL | HLA-B*5101
|
387.200
|
| polymerase PB2 | 759 | QBP33423.1 | 142-150 | RRVDINPGH | HLA-B*2705
|
9000.000
|
| hemagglutinin | 560 | QBP33427.1 | 316-324 | YIGVKSLKL | DRB1-0703
|
72.41
|
| polymerase PB2 | 759 | QBP33423.1 | 732-740 | LVMKRKRDS | DRB1-1301
|
89.77
|
| polymerase PB2 | 759 | QBP33423.1 | 730-738 | VVLVMKRKR | DRB1-1328
|
59.09
|
| polymerase PB2 | 759 | QBP33423.1 | 210-218 | LVRKTRFLP | DRB1-1327
|
55.68
|
Toxicity prediction: ToxinPred (Gupta et al., 2013) used for toxicity prediction of selected T- cell epitopes. ToxinPred tool is a unique in-silico method based on Support Vector Machine (SVM) in predicting toxicity of peptides along with important physico-chemical properties viz Charge, Hydrophobicity, Hydropathicity, Hydrophilicity and Molecular weight. The selected epitopes were subjected to ToxinPred and only non-toxic T-cell epitopes were selected for further studies (Table 5).
Table 5. Toxicity prediction of the peptides by ToxinPred
| PEPTIDE SEQUENCE | SVM SCORE | PREDICTION | HYDROPHOBICITY | HYDROPATHICITY | HYDROPHILICITY | CHARGE | MOL WT |
| FQGRGVFEL | -1.37 | NON-TOXIN | -0.05 | 0.14 | -0.23 | 0.00 | 1052.33 |
| AEIEDLIFL | -0.94 | NON-TOXIN | 0.16 | 1.19 | -0.13 | -3.00 | 1062.36 |
| IIEGRDRTL | -0.94 | NON-TOXIN | -0.32 | -0.48 | 0.69 | 0.00 | 1072.36 |
| RRVDINPGH | -0.91 | NON-TOXIN | -0.44 | -0.39 | 0.60 | 1.50 | 1063.31 |
| YIGVKSLKL | -1.14 | NON-TOXIN | 0.01 | 0.67 | -0.32 | 2.00 | 1020.42 |
| LVMKRKRDS | -0.84 | NON-TOXIN | -0.60 | -1.24 | 1.19 | 3.00 | 1132.51 |
| VVLVMKRKR | -0.71 | NON-TOXIN | -0.37 | 0.17 | 0.49 | 4.00 | 1128.62 |
| LVRKTRFLP | -0.65 | NON-TOXIN | -0.30 | -0.07 | 0.11 | 3.00 | 1129.54 |
Molecular Docking: 3D structures of selected epitopes were predicted by PEPstrMOD while HPEPDOCK Server was employed to generate homology model of alleles. Template PDB ID (protein data bank) formed by server was used for alleles model (table 6). HPEPDOCK Server has been utilized to perform docking study of epitopes with alleles models (Figure 1-8). The best conformation of docked complex was chosen on the basis of minimum docking score (table 7).
Table 6. Template PDB ID for modeling of selected HLA alleles
| S.No. | Allele | Template of model | Sequence Identity |
| 1 | HLA-B*2705 | 6AT5 A | 92.8% |
| 2 | DRB1*1328 | 6ATF B | 92.1% |
| 3 | HLA-B*0705 | 6AT5 A | 99.4% |
| 4 | HLA-B*5101 | 6AT5 A | 90.9%, |
| 5 | DRB1*0703 | 4H25 B | 84.3% |
| 6 | DRB1*1301 | 6PX6 B | 66.3% |
| 7 | DRB1*1327 | 6PX6 B | 66.3%, |
Figure 1: Docked complex of Nucleocapsid protein epitope AEIEDLIFL& HLA-B*2705 allele
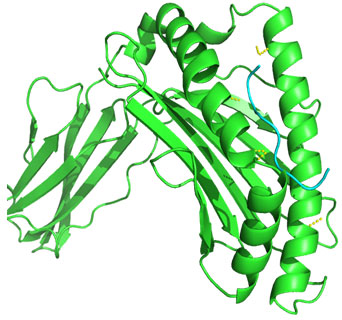
Figure 2: Docked complex of Nucleocapsid protein epitope FQGRGVFEL& HLA-B*0705 allele
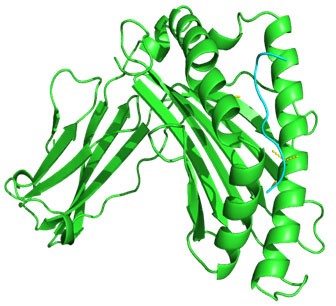
Figure 3: Docked complex of polymerase PAprotein epitope IIEGRDRTL & HLA-B*5101allele
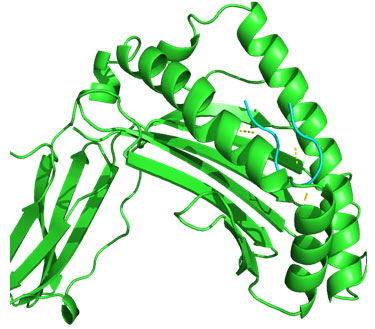
Figure 4: Docked complex of polymerase PB2 protein epitope LVMKRKRDS & DRB1-1301 allele
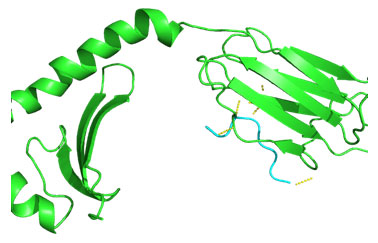
Figure 5: Docked complex of polymerase PB2 protein epitope LVRKTRFLP & DRB1-1327 allele
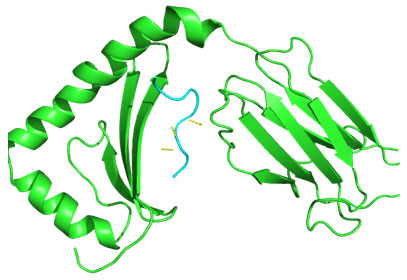
Figure 6: Docked complex of polymerase PB2 protein epitope RRVDINPGH & HLA-B*2705 allele
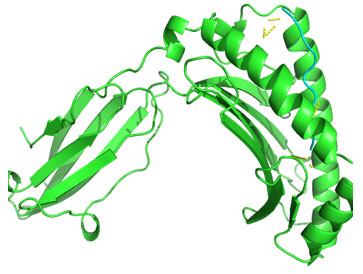
Figure 7: Docked complex of polymerase PB2 protein epitope VVLVMKRKR &DRB1-1328 allele
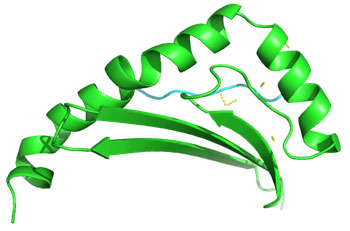
Figure 8: Docked complex of hemagglutininprotein epitope YIGVKSLKL&DRB1-0703 allele

Docking of selected nanomer T-cell epitopes FQGRGVFEL, AEIEDLIFL, IIEGRDRTL, RRVDINPGH, YIGVKSLKL, LVMKRKRDS, VVLVMKRKR, LVRKTRFLP with their corresponding allele HLA-B*0705, HLA-B*2705, HLA-B*5101, HLA-B*2705, DRB1-0703, DRB1-1301, DRB1-1328, DRB1-1327 respectively showed stable HLA–peptide complexes with docking score -229.708, -184.637, -206.640, -197.206, -176.581, -159.444, -177.159, -227.036 respectively (Table 7).
Table 7. Docking result of selected T-cell epitopes with allele structures.
| S.No. | Protein name | Epitopes | HLA class alleles | Docking Score |
| 1 | nucleocapsid protein | FQGRGVFEL | HLA-B*0705 | -229.708 |
| 2 | nucleocapsid protein | AEIEDLIFL | HLA-B*2705 | -184.637 |
| 3 | polymerase PA | IIEGRDRTL | HLA-B*5101 | -206.640 |
| 4 | polymerase PB2 | RRVDINPGH | HLA-B*2705 | -197.206 |
| 5 | hemagglutinin | YIGVKSLKL | DRB1-0703 | -176.581 |
| 6 | polymerase PB2 | LVMKRKRDS | DRB1-1301 | -159.444 |
| 7 | polymerase PB2 | VVLVMKRKR | DRB1-1328 | -177.159 |
| 8 | polymerase PB2 | LVRKTRFLP | DRB1-1327 | -227.036 |
Epitope antigenicity determination: VaxiJen is used with default parameters to predict the antigenicity of epitopes as vaccines candidate. All the antigenic epitopes with their respective predicted score (value greater than 01) were selected (table 8 & 9).
Table 8. Vaxijen for HLA class-I epitopes
| S. No. | SEQUENCE | VAXIJEN RESULT |
| 1 | FQGRGVFEL | 1.2783 (PROBABLE ANTIGEN) |
| 2 | AEIEDLIFL | 1.0317 (PROBABLE ANTIGEN) |
| 3 | IIEGRDRTL | 1.2600 (PROBABLE ANTIGEN) |
| 4 | RRVDINPGH | 2.5888 (PROBABLE ANTIGEN) |
Table 9. Vaxijen for HLA class-II epitopes
| S. No. | SEQUENCE | VEXIJEN RESULT |
| 1 | YIGVKSLKL | 1.9299 (PROBABLE ANTIGEN) |
| 2 | LVMKRKRDS | 1.9837 (PROBABLE ANTIGEN) |
| 3 | VVLVMKRKR | 2.4990 (PROBABLE ANTIGEN) |
| 4 | LVRKTRFLP | 1.6827 (PROBABLE ANTIGEN) |
Immunogenicity prediction of selected epitopes: Immunogenicity of nanomer HLA class I selected epitopes were analysed by IEDB. The selected epitopes with positive value showed high immunogenicity (Table 10).
Table 10. Immunogenicity of HLA class I epitopes
| S.NO | SEQUENCE | EPITOPE LENGTH | IMMUNOGENIGITY SCORE |
| 1 | FQGRGVFEL | 9 | 0.29224 |
| 2 | AEIEDLIFL | 9 | 0.33583 |
| 3 | IIEGRDRTL | 9 | 0.20424 |
| 4 | RRVDINPGH | 9 | 0.16967 |
The selected T-cell epitopes FQGRGVFEL, AEIEDLIFL, IIEGRDRTL, RRVDINPGH, YIGVKSLKL, LVMKRKRDS, VVLVMKRKR, LVRKTRFLP also show positive values of antigenicity & immunogenicity (in case of HLA class I) as shown in table 8-10. We have previously published similar work for HLA class I alleles for Nipah and HLA class II alleles for Hendra viruses (Kamthania and Sharma, 2015; Kamthania et al., 2019).
CONCLUSION
In this study, we have identified the potential nanomer T-Cell epitopes as vaccine candidate against H9N2 virus. The results confirming high binding affinity of selected epitopes with HLA alleles, stable complex formation tendency with HLA allele and tendency to induce high and specific immunogenic response makes the selected nanomer T-Cell epitopes to be a potential candidate for epitope based vaccine development against H9N2 virus infection. Hence reported nanomer epitopes may undergo further in-vivo trials to develop vaccine against H9N2 virus infection.
ACKNOWLEDGEMENTS
The authors are grateful for the necessary computational facilities and constant support provided by the faculty members of Department of Biotechnology, Faculty of Life Sciences, IAMR, Ghaziabad, India.
Conflict of interest: Authors declares that there is no conflict of interest.
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
REFERENCES
Ali, M., Yaqub, T., Mukhtar, N., Imran, M., Ghafoor, A., Shahid, M.F., Naeem, M., Iqbal, M., Smith, G.J. and Su, Y.C. (2019) Avian influenza A (H9N2) virus in poultry worker, Pakistan, 2015. Emerging Infectious Diseases, 25(1), p.136.
Butt, K.M., Smith, G.J., Chen, H., Zhang, L.J., Leung, Y.C., Xu, K.M., Lim, W., Webster, R.G., Yuen, K.Y., Peiris, J.M. and Guan, Y. (2005) Human infection with an avian H9N2 influenza A virus in Hong Kong in 2003. Journal of clinical microbiology, 43(11), pp.5760-5767.
Calis, J.J., Maybeno, M., Greenbaum, J.A., Weiskopf, D., De Silva, A.D., Sette, A., Keşmir, C. and Peters, B. (2013) Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol, 9(10), p.e1003266.
Doytchinova, I.A. and Flower, D.R. (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC bioinformatics, 8(1), p.4.
Geourjon, C. and Deleage, G. (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics, 11(6), pp.681-684.
Gupta, S., Kapoor, P., Chaudhary, K., Gautam, A., Kumar, R., Raghava, G.P. and Open Source Drug Discovery Consortium, (2013) In silico approach for predicting toxicity of peptides and proteins. PloS one, 8(9), p.e73957.
Kamthania, M. and Sharma, D.K. (2015) Screening and structure-based modeling of T-cell epitopes of Nipah virus proteome: an immunoinformatic approach for designing peptide-based vaccine. 3 Biotech, 5(6), pp.877-882.
Kamthania, M., Srivastava, S., Desai, M., Jain, A., Shrivastav, A. and Sharma, D.K. (2019) Immunoinformatics Approach to Design T-cell Epitope-Based Vaccine Against Hendra Virus. International Journal of Peptide Research and Therapeutics, 25(4), pp.1627-1637.
Killingley, B. and Nguyen‐Van‐Tam, J. (2013) Routes of influenza transmission. Influenza and other respiratory viruses, 7, pp.42-51.
Li, R., Adel, A., Bohlin, J., Lundkvist, Å., Olsen, B., Pettersson, J. H. O., & Naguib, M. M. (2020). Phylogeographic dynamics of influenza A (H9N2) virus crossing Egypt. Frontiers in microbiology, 11, p.392.
Pan, Y., Cui, S., Sun, Y., Zhang, X., Ma, C., Shi, W., Peng, X., Lu, G., Zhang, D., Liu, Y. and Wu, S. (2018) Human infection with H9N2 avian influenza in northern China. Clinical Microbiology and Infection, 24(3), pp.321-323.
Potdar, V., Hinge, D., Satav, A., Simões, E.F., Yadav, P.D. and Chadha, M.S. (2019) Laboratory-confirmed avian influenza a (H9N2) virus infection, India, 2019. Emerging infectious diseases, 25(12), p.2328.
Pusch, E.A. and Suarez, D.L. (2018) The multifaceted zoonotic risk of H9N2 avian influenza. Veterinary sciences, 5(4), p.82.
Robinson, J., Halliwell, J.A., McWilliam, H., Lopez, R., Parham, P. and Marsh, S.G. (2012) The imgt/hla database. Nucleic acids research, 41(D1), pp.D1222-D1227.
Shanmuganatham, K., Feeroz, M.M., Jones-Engel, L., Smith, G.J., Fourment, M., Walker, D., McClenaghan, L., Alam, S.R., Hasan, M.K., Seiler, P. and Franks, J. (2013) Antigenic and molecular characterization of avian influenza A (H9N2) viruses, Bangladesh. Emerging infectious diseases, 19(9), p.1393.
Singh, H. and Raghava, G.P.S. (2001) ProPred: prediction of HLA-DR binding sites. Bioinformatics, 17(12), pp.1236-1237.
Singh, S., Singh, H., Tuknait, A., Chaudhary, K., Singh, B., Kumaran, S. and Raghava, G.P. (2015) PEPstrMOD: structure prediction of peptides containing natural, non-natural and modified residues. Biology direct, 10(1), p.73.
Zhou, P., Jin, B., Li, H. and Huang, S.Y. (2018) HPEPDOCK: a web server for blind peptide–protein docking based on a hierarchical algorithm. Nucleic acids research, 46(W1), pp.W443-W450.

