1Biological Science Dept., Faculty of Sciences, King Abdul-Aziz Univ., KSA
2Princess Doctor Najla Bint Saud Al Saud Distinguished Research Centre for Biotechnology Jeddah, Saudi Arabia
3Biology Department, Faculty of Science, Taif University, Taif, Zip Code 21974, P.B 888, KSA
4Cell Biology Department National Research Center, Dokki, Giza Egypt.
5Microbial Genetics Department Genetic Engineering & Biotechnology Division National Research Center, Dokki, 12622 Giza Egypt
Corresponding author email: saboaba@kau.edu.sa
Article Publishing History
Received: 21/10/2020
Accepted After Revision: 10/12/2020
36 Bacillus native strains were isolated and identified using 16s gene sequencing analysis. Universal primer of 16s rRNA gene was used and PCR amplifications were done with all DNA isolation from all isolated strains. Mixture of non-identical 16S rRNA amplicons products produced and bioinformatics data analysis of all yielded variable consensus sequence were done using different software programs. The data analysis of sequences displayed some sequence variability occurs among the multiple 16S rRNA genes. Bioinformatical analysis revealed that the nucleotide sequences obtained from the isolates were different homologous to the known DNA sequence using NCBI Blast software. Results revealed that the obtained Bacillus isolated strains were belonging to17 different species. No noticeable base frequencies variation were observed in base frequencies for all the 39 isolates except B.simplex strains in in cytosine base. Results of phylogenic analysis revealed that all isolated strains are clustered analysis in 2 large clusters. The Polymorphism and Genetic Diversity among the isolates were also done using Clustalx and Maft build analysis, the monomorphic and polymorphic sits of all strains also analyzed.Further study for complete genome sequence analysis will be needed. The bioinformatics tools were succeeded in analyze all our native bacillus strains.
Bacillus. DNA, 16s rRNA, Genes, Bioinformatics Analysis.
Hadadi F. M, Bataweel N, Ali H. S. H. M, Alghamdi K. M, Sabry A, Ganash M, Abo-Aba S. E. M. Identification and Bioinformatic Analysis of 16s rRNA Gene Sequences of Native Bacillus sp.isolated Strains from Saudi Arabia. Biosc.Biotech.Res.Comm. 2020;13(4).
Hadadi F. M, Bataweel N, Ali H. S. H. M, Alghamdi K. M, Sabry A, Ganash M, Abo-Aba S. E. M. Identification and Bioinformatic Analysis of 16s rRNA Gene Sequences of Native Bacillus sp.isolated Strains from Saudi Arabia. Biosc.Biotech.Res.Comm. 2020;13(4). Available from: <a href=”https://bit.ly/3n0zgic”>https://bit.ly/3n0zgic</a>
Copyright © Hadadi et al., This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) https://creativecommns.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
The microbes in the rhizosphere are a diverse mixture of microorganisms that can actively interact with the host plant in different ways. The studies on the microbial rhizosphere communities have shown the significant influence of plant species and cultivars in shaping microbial communities in the rhizosphere (Lei et al., 2018).Soil is an important environment and contain all number of additives such as mineral with organic elements for plant growth, also contain huge number of microorganisms safe and non-safe for plant (Wołejko et al., 2020). The soil conditions are important for plant and microorganisms such as physical respectabilities and chemical constituents (Tajik et al., 2020).
Many microorganisms isolated and identified from soil all over the world by scientist’s, such as bacteria, algae, fungi, etc. all these microorganisms are benefits for plant production and, also fight plant pathogens. So, it plays a main role for biological control of plant pathogens. Significant characters from soil bacteria such as gibberellic acid (GA3), indole-3-acetic acid (IAA) which are phytohormones, nitrogen fixation, and nutrient solubilization show main factors in plants production (Tabatabaei,et al., 2016, Wołejko et al., 2020).
Bacillus strains have been extensively studied specially Bacillus thuringiensis because of its huge benefits and safe for human being. There are too many applications of these bacillus under study in biological control because it used for fight many types of insects (Sanahuja,et al., 2011). B. amyloliquefaciens, B. subtilis, B. megaterium and B. thuringiensis and other similar bacterial strains isolated from soil types when microbiome in soil studied by (Boottanun et al., 2017; Kui et al., 2019; Panneerselvam et al., 2019). Isolation of bacillus strains and species from soil is easy way, it isolated from various depth (Labo et al., 2018: Kui et al., 2019).
The 16S rRNA gene sequence is used for phylogenetic studies based on the produced sequences (Woese et al., 1977). Bacterial 16S rRNA genes are located within the rRNA operons, which also contain genes for 23S rRNA, 5S rRNA, tRNA, and associated intergenic spacer regions (Woese et al., 1990). Identification of bacterial strains to the genus and species level is preferred according to sequencing of rRNA genes because this gene contain highly conserved and hypervariable regions (Anda et al., 2015). 16S rRNA sequence analysis used for identification of bacterial isolates this method is reliable and fast than classical conventional methods. (Woo et al., 2008). Phenotypic characteristics analysis including Gram staining, cell morphology, and biochemical properties are still the first steps in species identification, and 16S rRNA sequence analysis may be performed only when the phenotypic results are not definitive (Clarridge. 2004).
In the genome of some single strains there are variation in 16S rRNA gene sequences such as the presence of single nucleotide polymorphisms (SNPs) or small insertions or deletions among the multiple 16S rRNA copies (Sacchi et al., 2002;Acinaset al., 2004). By universal 16S rRNA designed primers to target conserved sites and targeted the 16S rRNA gene in isolated DNA from any bacterial strain and sequencing of such amplified products, now useful method for bacterial identification (Petti,et al., 2015). In this study isolation and molecular identification of some bacillus strains from Saudi Arabia soil were done. Bioinformatics analysis and comparison of multiple 16S rRNA genes sequencing from isolated bacterial strains were done.
MATERIAL AND METHODS
Four different soil samples were collected from different regions of Jeddah City. Isolation of Bacillus strains were done. 0.5g of soil samples was suspended in 10 ml of sterile distilled water, soil samples was homogenized vigorously with vortex. After mixing 1 ml of the supernatant was stored at80oC for some minutes to kill most non-spore-forming bacteria.100 µL of suspension spread over nutrient agar plates. The apparent colonies were spread on the entire surface of nutrient agar m plates with triplicate. After incubation at 30oC for two days all colonies like bacillus colonies were chosen. The obtained single colony re-purified by re-streaking over Nutrient agar plates several times, stored in Nutrient agar slants at 4oC, and long term storage in Nutrient agar broth with 30% glycerol at -80oC (Masoud et al., 2015; Al-Yahyawy et al., 2019).
Extraction of DNA: Extraction of DNAs was carried out by I mL of each isolated strains was centrifuged to collect the pellet. 200µL of TES buffer (50mM EDTA, 100mM Tris pH8, and 10% SDS) and 20µL Lysozyme added to the bacterial pellet with gentle pipetting at 37oCfor 60 min., 20µL of proteinase K solution added to the mix incubated at 37oC for 60 min., the pellet suspension transferred to ice path for 5 min. then 250 µL of Sodium acetate 4 Madded. The mix was spin down for 5 min. at 10.000 rpm for 5 min. clear supernatant was transferred to a new tube and 250 µL of chloroform/isoamyle alcohol added to the supernatant in ice path for 5 min. centrifugation was done at 10.000 rpm for 5 min. the collected DNA pellet dried for 10
min in laminar flow cabinet. 50 µL of TE (10mM Tris pH8, 1mM EDTA) added to each tube. The DNA were detected by electrophoresis in 1.5% agarose gel (Sabir, et al., 2013; Al-Yahyawy,et al., 2019.).
16s rDNA isolation and analysis: PCR amplification and sequencing of 16S rDNA16S gene sequencing and analysiswere done. The amplified PCR products of the 16S rRNA gene bacterial gene fragments were detected by electrophoresis in 1.5% agarose gel. The amplified fragments were purified and sequenced at MACROGEN sequencing company, Seoul, Korea using the automated sequencer ABI 3100 (Applied Biosystems) with Big Dye Terminator Kit v. 3.1 (Applied Biosystems). Primers 518 F (5’ C CA GCAG CC GC GG TAATACG – 3′) and 800 R (5′- TA CC AG GG TA TCTA AT CC -3′) were used for sequencing. The sequences obtained were edited with the software Vector NTI Suite 9, and compared with the NCBI database through BLAST searches. In this comparison, sequences of type strains most closely related to the sequences of the isolates were searched. For the definition of operational taxonomic units (OTUs), a similarity limit of 97% was adopted (Sabir, et al., 2013; Al-Yahyawy,et al., 2019).
Bioinformatics and data analysis: All the 39 sequences were subjected to ncbi BLAST search tool http://blast.ncbi.nlm.nih.gov to detect non-chance sequence similarity. BLAST search was restricted to 16S ribosomal RNA (Bacteria and Archaea), where models (XM/XP) as well as unclultured/environmental samples were also filtered out, such that more reliable results would be attained. Each individual sequence was solely blastd, where blast hit with the lowest expect-value (which indicate number of non-chance alignments) was picked. In order to ensure that Blast out puts were governed by expected-value (aka e-value), Blast algorithm parameter was decreased such the expected threshold was set to more stringent value of 1e−6. Alignment of the 39 sequence was carried out using version 2 of Clustalx (Larkin et al., 2007)
Exploratory data and phylogenetic analyses were carried out under R Project for Statistical Computing (R Core Team, 2017). Where Exploratory data analysis was done using Seqinr (Charif&Lobry, 2007) R package. Phylogenetic analysis was carried out by ape package (Paradis, et al., 2004). Reconstruction of the phylogenetic tree was done using Neighbor joint method (Nei, 1987).DnaSP (LibradoandRozas, 2009) software was used to analyze the haplotype diversity (Hd), the average number of nucleotide differences, the average number of nucleotide differences (Tajima, 1983), the nucleotide diversity (π). The polymorphic site (S), the singleton variable sites (SP), and the parsimony informative sites (PIP) for each gene, and the average number of nucleotide substitutions per site between species (Dxy) (Lynch and Crease, 1990).
RESULTS AND DISCUSSION
Bacterial samples: Out of 36 Bacillus bacteria from soil samples near Jeddah were isolated on nutrient agar plates as pure culture in this study(Table 1) and fig. (1).The isolated strains were re-purified and streaked over the same plates (Fig. 1). 16s rDNA gene of the isolates were done as shown in fig (2). The16S rRNA were identified by 16S rRNA sequence analysis at Macrogen Company Korea, with Macrogen universal primer as identified in materials and methods. The results of 16 s gene isolation as shown pure 16S rRNA gene isolation in fig (2).
Figure 1: An example for purification of Bacillus isolates appeared over nutrient agar plates.

Table 1. Soil Samples Locations
| Sample number | city | location |
| 1 | Tabuk | 28.378089,36.565889 |
| 2 | Baljurashi | 19.865853,41.582135 |
| 3 | Khobar | 26.213500,50.188347 |
| 4 | Jeddah | 21.490086, 39.202683 |
Figure 2: 1 16S rRNA isolation on agarose gel electrophoresis

NCBI BLAST query: BLAST results are shown in Table (2) for all the 39 isolates. The criteria used for query sequence aimed to narrow down the search space (database), as the smaller the database the more likely to contain sequence of interest. For all the 39 queries zero E-values were attained indicated that all alignments were non-chance alignments. The query cover percentage ranged from 93 to 100 % with the only one exception for isolate 33. The vast majority of values of identical percent (% of how similar the query sequence is to the target sequence) ranged from 97% to 100% with only 2 isolates have identical percent of 80% and 82% table( 2).
The distribution of isolates/species/distribution is presented graphically in Figure (3)the NCBI BLAST query resulted in 17 species, mostly belong to Bacillus sp,namely Species number 11 B.paramycoides, 4 B.oceanisediminis, 3 B.wiedmannii, 2 for each of B.aryabhattai, B.drentensis, B.megaterium, B.niacini, B.simplex, Lysinibacillusfusiformis and Lysinibacillusmacroides, where only one isolate was found to be belong to each of B.firmus, B.onubensis,B.persicus,B.thioparans,B.toyonensis,Lysinibacilluspakistanensis and Oceanobacilluslongus.
Figure 3: The distribution of isolated bacterial strains at species level
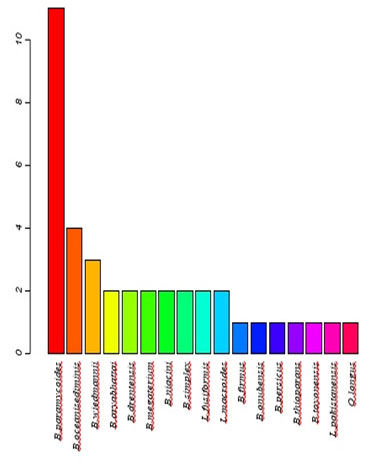
Table 2. blast results of isolated Bacillus strains
| Isolate | BLAST Results
Species |
Query cover% | E-value | %
identical |
| 11-27F | B.paramycoides | 100 | 0.00 | 99 |
| 12-27F | B.paramycoides | 100 | 0.00 | 100 |
| 13-27F | B.paramycoides | 99 | 0.00 | 100 |
| 14-27F | B.paramycoides | 100 | 0.00 | 99 |
| 15-27F | B.paramycoides | 100 | 0.00 | 99 |
| 27-27F | B.paramycoides | 99 | 0.00 | 100 |
| 32-27F | B.paramycoides | 100 | 0.00 | 100 |
| 34-27F | B.paramycoides | 99 | 0.00 | 100 |
| 36-27F | B.paramycoides | 98 | 0.00 | 99 |
| 46-27F | B.paramycoides | 99 | 0.00 | 100 |
| 6-27F | B.paramycoides | 99 | 0.00 | 100 |
| 30-27F | B.oceanisediminis | 95 | 0.00 | 99 |
| 37-27F | B.oceanisediminis | 93 | 0.00 | 99 |
| 49-27F | B.oceanisediminis | 95 | 0.00 | 99 |
| 29-27F | B.oceanisediminis | 96 | 0.00 | 99 |
| C2-27F | B.wiedmannii | 99 | 0.00 | 100 |
| 18-27F | B.wiedmannii | 99 | 0.00 | 100 |
| 10-27F | B.wiedmannii | 100 | 0.00 | 99 |
| 19-27F | B.aryabhattai | 99 | 0.00 | 100 |
| 20-27F | B.aryabhattai | 99 | 0.00 | 100 |
| 2-27F | B.megaterium | 99 | 0.00 | 100 |
| 8-27F | B.megaterium | 99 | 0.00 | 100 |
| 22-27F | B.simplex | 100 | 0.00 | 99 |
| 28-27F | B.simplex | 97 | <0.00 | 82 |
| 43-27F | B.niacini | 99 | 0.00 | 99 |
| 5-27F | B.niacini | 99 | 0.00 | 98 |
| 31-27F | B.drentensis | 100 | 0.00 | 99 |
| 4-27F | B.drentensis | 100 | 0.00 | 97 |
| 40-27F | Lysinibacillusfusiformis | 100 | 0.00 | 98 |
| 41-27F | Lysinibacillusfusiformis | 100 | 0.00 | 98 |
| 50-27F | Lysinibacillusmacroides | 99 | 0.00 | 99 |
| 52-27F | Lysinibacillusmacroides | 99 | 0.00 | 99 |
| 1-27F | B.onubensis | 95 | 0.00 | 99 |
| 44-27F | B.firmus | 100 | 0.00 | 97 |
| 45-27F | B.thioparans | 99 | 0.00 | 98 |
| 48-27F | Lysinibacilluspakistanensis | 99 | 0.00 | 96 |
| 9-27F | B. persicus | 99 | 0.00 | 99 |
| C1-27F | B. toyonensis | 99 | 0.00 | 99 |
| 33-27F | Oceanobacilluslongus | 72 | <0.00 | 80 |
Exploratory data analysis: Table (3) shows a description of the isolated species as well as their sequence length and the percentages of GC content of each isolate. Sequence length varied greatly among the 39 isolates ranged from 463 bp for B. oceanisediminis to 500 bp for B. onubensis. The percentage of GC content were found to be constant within species with no similar trend was observed for the Saudi isolates. Generally speaking, % GC ranged from 52 for B.megaterium to 56 for B.persicus. Base frequencies for all the 39 isolates is shown graphically in figure (4)No noticeable variation were observed in base frequencies for all the 39 isolate exceptfor B.simplex where this species has a noticeable access in cytosine base.
Table 3. The sequence length and percentage of GC ratio of isolated bacillus strains
| Species | sequence Length | GC% |
| 12-B. paramycoides | 474 | 0.53 |
| 15-B. paramycoides | 471 | 0.53 |
| 32-B. paramycoides | 476 | 0.53 |
| 46-B. paramycoides | 476 | 0.53 |
| 27-B. paramycoides | 467 | 0.54 |
| 13-B. paramycoides | 477 | 0.53 |
| 36-B. paramycoides | 476 | 0.53 |
| 6-B. paramycoides | 476 | 0.53 |
| 11-B. paramycoides | 479 | 0.53 |
| 14-B. paramycoides | 478 | 0.53 |
| 34-B. paramycoides | 475 | 0.53 |
| 29-B. oceanisediminis | 463 | 0.55 |
| 30-B. oceanisediminis | 472 | 0.55 |
| 49-B. oceanisediminis | 467 | 0.55 |
| 37-B. oceanisediminis | 480 | 0.55 |
| 10-B. wiedmannii | 483 | 0.53 |
| 18-B. wiedmannii | 477 | 0.53 |
| C2-B. wiedmannii | 476 | 0.53 |
| 2-B. megaterium | 475 | 0.52 |
| 8-B. megaterium | 477 | 0.52 |
| 40-L. fusiformis | 479 | 0.53 |
| 41-L. fusiformis | 480 | 0.53 |
| 50-L. macroides | 475 | 0.53 |
| 52-L. macroides | 468 | 0.53 |
| 48-L. pakistanensis | 481 | 0.52 |
| 19-B. aryabhattai | 474 | 0.53 |
| 20-B. aryabhattai | 474 | 0.53 |
| 43-B. niacini | 473 | 0.55 |
| 5-B. niacini | 476 | 0.55 |
| 4-B. drentensis | 481 | 0.55 |
| 31-B. drentensis | 466 | 0.56 |
| 22-B. simplex | 474 | 0.54 |
| 28-B. simplex | 470 | 0.52 |
| 44-B. firmus | 478 | 0.55 |
| 45-B. thioparans | 476 | 0.54 |
| 1-B. onubensis | 500 | 0.52 |
| 33-O. longus | 499 | 0.52 |
| C1-B. toyonensis | 471 | 0.54 |
| 9-B. persicus | 474 | 0.56 |
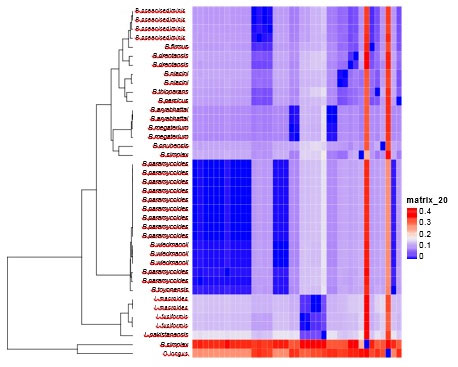
Cluster & phylogenetic analysis: Cluster analysis and heatmap were carried out as pre-processing step to glean an insight into the data distribution. Results of cluster analysis are shown graphically in figure (5), where 2 large clusters are shown. The first cluster comprises merely 2 species B.simples and Oceanobacilluslongus, which indeicate that these two speices are distantly densely related to the rest of species. All other 37 species were clurested together in the second cluster occupying different clades.
Figure 4: Base frequencies for all the 39 isolates bacillus isolated strains
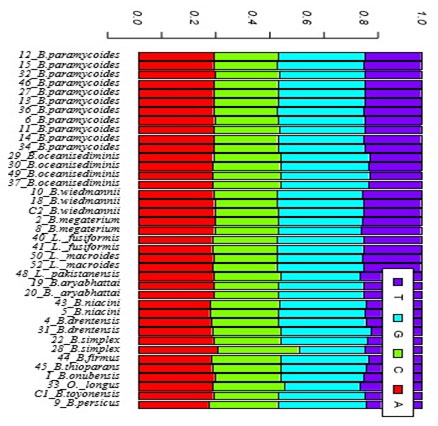
Table 4. Length of conserved regions, conservation, homzigosity and P-valuesof 39 isolates
| Region Start-End | Conservation | Homozigosity | P-value |
| 61-138 | 0.61 | 0.1 | 0.04 |
| 63-140 | 0.61 | 0.9 | 0.04 |
| 65-157 | 0.60 | 0.9 | 0.02 |
| 82-160 | 0.61 | 0.9 | 0.03 |
| 209-290 | 0.60 | 0.95 | 0.04 |
| 244-344 | 0.60 | 0.97 | 0.02 |
| 271-393 | 0.60 | 0.97 | 0.01 |
Figure 5: Cluster analysis of 36 isolated bacillus strains
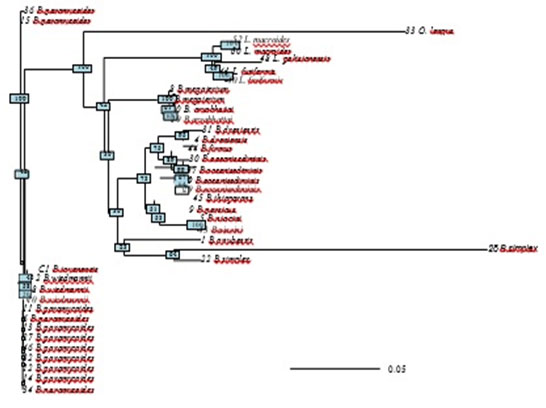
Evolutionary relationships of taxa was inferred by reconstructing phylogenetic tree using the Neighbor-Joining method (Saitou & Nei, 1987). The optimal tree with the sum of branch length = 1.2 is shown (figure 6). The results of phylogenetic analysis deeply sports the findings of cluster analysis. B.simples and Oceanobacillus longus were found to have the longest branches of the phylogenetic tree, which again indicate that these two species are distantly densely related to the rest of species. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method are in the units of the number of base substitutions per site. The analysis involved 39 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 437 positions in the final dataset.
Figure 6: Phylogenetic tree using the Neighbor-Joining method Maximum Likelihood method indicated and reconstructing the evolutionary relationships of isolated bacillus strains.
Polymorphism and Genetic Diversity among isolated bacillus strains: All 39 isolates were aligned using both Clustalx and Maft build in ape package (Paradis et al., 2004). As the alignment results of the two software were found to be identical, the robustness of the alignment method is ensured. General information about the polymorphisms on the isolates is found in a table (4). The number of sites was 442, of which 233 sites were fond monomorphic, 209 sites were polymorphic. These polymorphic sites divided into 104 parsimony informative sites (i.e. sites that have a minimum of two nucleotides that are present at least twice) and 105 singleton variable sites (non- informative).In the present data set number of haplotypes were 22 where haplotype diversity was 0.94±0.03 Nucleotide diversity (π) is defined as the average number of nucleotide differences per site between two sequences Nei (1987). Estimate of nucleotide diversity was found to be 0.10, where the average number of nucleotide differences was 44 (table 5).
Table 4. Estimated parameters of the polymorphic sites for 39 bacillus isolated strains
| Isolate No. | No. Mono. | No. Poly | Parimony | Singleton |
| Sites | -morphic
sites |
-morphic
sites |
informative
sites |
variable
sites |
| 442 | 233 | 209 | 104 | 105 |
Table 5. Estimated parameters of DNA polymorphism for 39 bacillus isolated strain

Table (6) shows the conserved regions along the 39 isolates and measurements of conservation (C), homosigoisty and P- value. Conservation (C) is calculated as the proportion of conserved sites in the alignment region, where homosigosity is measured as 1- heterzygosity. A total of seven conserved regions were observed, 6 of which are overlapping. The P-value for 6 of these seven con- served region were 0.05, where the P-value for the seventh conserved region was0.01
Table 6. Length of conserved regions, conservation, homzigosity and P-values of all 39 bacillus isolated strains
| Region Start-End | Conservation | Homozigosity | P-value |
| 61-138 | 0.61 | 0.1 | 0.04 |
| 63-140 | 0.61 | 0.9 | 0.04 |
| 65-157 | 0.60 | 0.9 | 0.02 |
| 82-160 | 0.61 | 0.9 | 0.03 |
| 209-290 | 0.60 | 0.95 | 0.04 |
| 244-344 | 0.60 | 0.97 | 0.02 |
| 271-393 | 0.60 | 0.97 | 0.01 |
16S rRNA gene sequencing has been a widely used method for the identification of bacterial isolates from different environmental samples. The traditional culture methods for identification depending on phenotypic characteristics and biochemical testing. These methods require minimum equipment but differentiate only between common species (Springer, et al., 2001; Tortoli, et al, 2001). The 16S rRNA gene sequencing has been described and available technique for bacterial identification (Mauti, et al., 2019). The timing needed for 16S rRNA gene sequencing confirmd as speed and accuracy for identification of bacterial isolates comparing to normal traditional identification methods (Cloud, et al., 2002; Kirschner,et al., 2000; Turenne, et al., 2001; Sabir, et al., 2013; Al-Yahyawyet al., 2019).
Comparing of microbial communities with the sequenceswith closely related species in the similar 16s r RNA sequences of databases help all scientists all over the world to identify the bacterial species in their samples (Burns, et al., 2015; Claesson, et al., 2010). The 16S rRNA gene become more commonly used for rapid identification of unknown isolates environmental samples and for confirmation of variant sequence data that might have been generated by the higher-throughput methods (Clarridge 2004). The number of 16S rRNA gene sequences in different databases is increased daily (Boudewijns, et al., 2006). Our results confirmed that a description of the isolated species as well as their sequence length and the percentages of GC content of each isolate these data are inagreement with literatures (Turenne, et al., 2001; Sabir, et al., 2013; Al-Yahyawyet al., 2019).
No noticeable variation in the base frequencies for all the 39 isolates except for B.simplex where this species has a noticeable access in cytosine base. Also obtained results shown all 37 species were clurested together in the second cluster occupying different clades (Dréan,et al., 2015).I n the present data set we show number of nucleotide diversity according to Nei (1987). A total of seven conserved regions were observed along the 39 isolates and measurements of conservation (C), homosigoisty and P- value. Conservation (C) is calculated as the proportion of conserved sites in the alignment region, where homosigosity is measured as 1- heterzygosity (Paradis,et al., 2004).
CONCLUSION
In conclusion, these preliminary results confirm the fact that species identification by bioinformatics analysis approach may be sufficient for analyzing cultured and uncultured bacterial communities from environmental samplesto species level in some cases. By the public databases it resolve wasting time by traditional bacterial identification methods, but should be confirmed by complementary DNA sequencing. High-quality DNA sequences with SNP analysis could increase the power of databases to facilitate bacterial identification and discrimination between closely related species (Uelze, et al., 2020).
REFERENCES
Acinas, S. G., Marcelino, L. A., Klepac-Ceraj, V., Polz. M. F. (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186:2629–2635.
Anda, M., Ohtsubo, Y., Okubo, T., Sugawara, M., Nagata, Y., Tsuda, M., Minamisawa.,K., Mitsui, H. (2015) Bacterial clade with the ribosomal RNA operon on a small plasmid rather than the chromosome. ProcNatlAcadSci USA 112:14343–14347.
Boottanun, P., Potisap, C., Hurdle, J.G. and Sermswan, R.W. (2017) Secondary metabolites from Bacillus amyloliquefaciens isolated from soil can kill Burkholderia pseudomallei. AMB Express, 7: 1-11.
Boudewijns, M., Bakkers, J. M., Sturm, P.D. J. and Melchers, W. J. S. (2006) 16S rRNA Gene Sequencing and the Routine Clinical Microbiology Laboratory: a Perfect Marriage? Journal of Clinical Microbiology. 3469–3470
Bravo, A., Gill, S.S. and Soberón, M. (2007) Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control, Toxicon 49 (4) (2007) 423–435.
Burns, K. N., Kluepfel, D. A., Strauss, S. L., Bokulich, N. A., Cantu, D., and Steenwerth, K. L. (2015) Vineyard soil bacterial diversity and composition revealed by 16S rRNA genes: differentiation by geographic features. Soil Biol. Biochem. 91, 232–247.
Charif, D., &Lobry, R. J. 2007. SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. Pages 207–232 of: Bastolla, U., Porto, M., Roman, H.E., &Vendruscolo, M. (eds), Structural approaches to sequence evolution: Molecules, networks, populations. Biological and Medical Physics, Biomedical Engineering. New York: Springer Verlag. ISBN: 978-3-540-35305-8.
Claesson, M. J., Wang, Q., O’Sullivan, O., Greene-Diniz, R., Cole, J. R., Ross, R. P., et al. (2010). Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38, 1–13.
Clarridge, J. E. (2004) Impact of 16S rRNA Gene Sequence Analysis for Identification of Bacteria on Clinical Microbiology and Infectious Diseases. Clinical Microbiology Reviews. 840–862
Cloud, J. L., H. Neal, R. Rosenberry, C. Y. Turenne, M. Jama, D. R. Hillyard, and K. C. Carroll. 2002. Identification of Mycobacterium spp. by using a commercial 16S ribosomal DNA sequencing kit and additional sequencing libraries. J. Clin. Microbiol. 40:400–406.
Dréan, P., McAuley, C. M., Moore, S.C.,Fegan, N. and Fox, E.M. (2015) Characterization of the spore-forming Bacillus cereus sensulato group and Clostridium perfringens bacteria isolated from the Australian dairy farm environment.BMC Microbiology. 15 (38): 1-10.
Kirschner, P., and Bottger. E. C. (2000) Species identification of mycobacteria using rDNA sequencing. Methods Mol. Biol. 101:349–361.
Kui, L., Sun, H., Lei, Q., Gao, W., Bao, L., Chen, Y., Jia, Z. (2019) Soil microbial community assemblage and its seasonal variability in alpine tree Lineecotone on the eastern Qinghai-Tibet Plateau. Soil Ecology Letters. 1: 33–41.
Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R.and McGettigan, P.A., McWilliam, H., Valentin, F., Wal- lace, I.M., Wilm, A., Lopez, R., Thompson, J.D., & Gibson, T.J., Higgins D.G. 2007. Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–2948.
Lei, S., Xu, X., Cheng, Z. Xiong, J., Ma, R., Zhang, L., Yang, X., Zhu, Y., Zhang, B. and Tian, B. (2018) Analysis of the community composition and bacterial diversity of the rhizosphere microbiome across different plant taxa. Microbiology. 1-10.
Librado, P., and Rozas, J. (2009) DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 25, 1451–1452.
Lobo, K.S., Soares-da-Silva, J., Cleoneide da Silva ,M., Tadeid, W.P., Polanczyke, R.A., Valéria Cristina SoaresPinheiro, V.C.(2018) Isolation and molecular characterization of Bacillus thuringiensis found in soils of the Cerrado region of Brazil, and their toxicity to Aedesaegypti larvae. RevistaBrasileira de Entomologia., 62:1-5.
Mauti, E. M., Mauti, G. O., Ouno, G. A. and Mabeya, P.M (2019) Molecular Identification of Soil Bacteria by 16srDNA Sequence. Journal of Natural Sciences Research. 3 (14): 51-58.
Nei, M. (1987) Molecular Evolutionary Genetics. New York: Columbia Univ. Press.
Othman Al-Yahyawy, Fatimah M. Hadadi, Khalid M.S.Al-Ghamdi, AymanSabry
and Salah E. M. Abo-Aba (2019) Phylogenetic Relationship and Genetic Diversity of Bacillus thuriengensis Isolated from Soil Samples, Jeddah, KSA. International Journal of Current Advanced Research. 2319-6505.
Panneerselvam, P., Senapati, A. Kumar, U., Laxuman Sharma, L., Lepcha, P., Prabhukarthikeyan, S.R., Jahan, A. et al (2019) Antagonistic and plant‑growth promoting novel Bacillus species from long‑term organic farming soils from Sikkim, India. Biotechnology.9 :1-12.
Paradis, E., Claude, J., &Strimmer, J. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics, 20, 289–290.
Petti, C. A., Polage C. R. and Schreckenberger P. (2015) The Role of 16S rRNA Gene Sequencing in Identification of Microorganisms Misidentified by Conventional Methods. J. Clin. Microbiol. 43(12): 6123–6125.
Pu, Y., Ma, T., Hou, Y. and Sun, M. (2016) Anentomopathogenic bacterium strain, Bacillus thuringiensis as a biological control agent against the red palm weevil, Rhynchophorus ferrugineus (Coleoptera:Curculionidae): Efficacy of Bacillus thuringiensis against Rhynchophorus ferrugineus. Pest Management Science.73:1494-1502.
R Core Team. (2017) R: A Language and Environment for Statistical Com- putting. R Foundation for Statistical Computing, Vienna, Austria.
Sabir, J.S., Abo-Aba, S. E. M., Said, M.M., Hussein, R. M., Al-Saud, N. and Mutwakil, M. (2013) Isolation, Identification and RAPD-PCR analysis of New Isolated Bacillus thuriengensis. Life Science Journal 10(2): 1352- 1361.
Sacchi ,C.T., Whitney, A. M,, Mayer, L.W., Morey, R., Steigerwalt, A., Boras, A., Weyant, R. S., Popovic. T. (2002) Sequencing of 16S rRNA gene: a rapid tool for identification of Bacillus anthracis. Emerg Infect Dis 8:1117–1123.
Saitou, N., and Nei, M. (1987) The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425.
Sanahuja, G., Banakar, R. Twyman, R. M., Capell, T. and Christou, T (2011) Bacillus thuringiensis: a century of research, development and commercial applications. Plant Biotechnology 9: 283–300.
Santos, M.S., Dias, N.P., Costa, L.L., De Bortoli, C. P., Souza, E. H., Santos, A.C., De Bortoli, S.A. and Polanczyk, R. A. (2019) Interactions of Bacillus thuringiensis strains for Plutellaxylo stella (L.) (Lepidoptera: Plutellidae) susceptibility. Journal of Invertebrate Pathology. 168: 1-3.
Springer, B., L. Stockman, K. Teschner, G. D. Roberts, and E. C. Bottger. (1996) Two-laboratory collaborative study on identification of mycobacteria: molecular versus phenotypic methods. J. Clin. Microbiol. 34:296–303.
Tabatabaei, S., Ehsanzadeh, P., Etesam, H., Alikhani, H. A. and Glick, B. R. (2016) Indole-3-acetic acid (IAA) producing Pseudomonas isolates inhibit seed germination and α-amylase activity in durum wheat (Triticum turgidum L.). Spanish Journal of Agricultural Research. 14(1), 802.
Tajik, S., Ayoubi, S. and Lorenz, N. (2020) Soil microbial communities affected by vegetation, topography and soil properties in a forest ecosystem. Applied Soil Ecology. 149:1-12.
Tortoli, E., Bartoloni, Bottger, A. E. C., Emler, S., Garzelli, C., Magliano, E., Mantella,A. Rastogi, N., Rindi, L., Scarparo, C. and Urbano, P. (2001) Burden of unidentifiable mycobacteria in a reference laboratory. J. Clin. Microbiol. 39:4058–4065.
Turenne, C. Y., Tschetter, L., Wolfe, J. and Kabani, A. (2001) Necessity of quality-controlled 16S rRNA gene sequence databases: identifying nontuberculous Mycobacterium species. J. Clin. Microbiol. 39:3637–3648.
Uelze, L., Grützke, J., Borowiak, M., Hammerl, J. A., Juraschek, K., Deneke, C., Tausch, S. H. and Malorny, B (2020) Typing methods based on whole genome sequencing data. One Health Outlook. 2:3.
Woese, C. R, Fox, G. E. (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74:5088–5090.
Woese, C. R., Kandler, O., Wheelis, M. L. (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA. 87:4576–4579.
Wołejko, E., Jabłońska-trypuć, A., Wydro, U.,Butarewicza, A. and Łozowicka, B. (2020) Soil biological activity as an indicator of soil pollution with pesticides – A review. Applied Soil Ecology. 147: 1-13.
Zghal, R.Z., Ghedira, K., Elleuch, J., Marwa Kharrat, M. and Tounsi, S. (2018) Genome sequence analysis of a novel Bacillus thuringiensis strain BLB406 active against Aedes aegypti larvae, a novel potential bioinsecticide. International Journal of Biological Macromolecules. 116:153-162.
Zhang, W., Crickmore, N., George, Z., Xie, L., He, Y.Q., Li, Y. , Tang, J.L., Tian, L., Wang, X. and Fang, X. (2012) Characterization of new highly mosquitocidal isolate of Bacillus thuringiensis –an alternative to Bti? J. Invertebr. Pathol. 109 (2012) 217–222.

