Department of Biological Science and Engineering, Maulana Azad National Institute of Technology, Bhopal (India).
Corresponding author email: rashiraizada@gmail.com
Article Publishing History
Received: 17/04/2021
Accepted After Revision: 25/06/2021
Chikungunya (CHIKV) and Dengue (DENV) are common arboviruses usually found in tropical countries. To understand the molecular evolution pattern and regulation of viral genome the codon usage analysis serve as an important tool. This study utilized the two important parameters Nucleotide composition (Nc) and Relative Synonymous Codon Usage (RSCU) of codon usage analysis for detailed understanding. We have processed and analyzed 382 sequence of DENV and 641 sequences of CHIKV for Nucleotide composition (Nc) and Relative Synonymous Codon Usage (RSCU). A comparative analysis of RSCU between both pathogens (DENV and CHIKV) and between vector and host i.e. Ades aegytpti and Homo sapiens was carried out. It is hypothesized because DENV and CHIKV both are sharing common host and vector therefore the findings of our study will provide a detailed insight towards an understanding of molecular evolution between the virus vector and its host.
The study suggests that the codon usage patterns of these viruses are a combination of coincidence and antagonism.The observations recorded indicate that the codon usage pattern of DENV and CHIKV is dominated by mutation pressure under the influence of natural selection from its hosts. In conclusion, the overall codon usage bias is low in DENV and CHIKV i.e. mutation pressure is playing a key role in evolution. To the best of our knowledge, this is the first report describing the comparative analysis of codon usage in CHIKV and DENV genomes. The findings from this study are expected to increase our understanding of factors involved in viral evolution, and fitness towards hosts and the environment.
Dengue Virus, Chikungunya Virus, Codon Usage Bias, Mutational Pressure, Natural Selection
Raizada R, Pandey K. M . Comparative Analysis of Codon Usage of Dengue and Chikungunya Viruses with Human Host and Vector
Raizada R, Pandey K. M . Comparative Analysis of Codon Usage of Dengue and Chikungunya Viruses with Human Host and Vector. Biosc.Biotech.Res.Comm. 2021;14(2). Available from: <a href=”https://bit.ly/2StChgE“>https://bit.ly/2StChgE</a>
Copyright © Raizada and Pandey This is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) https://creativecommns.org/licenses/by/4.0/, which permits unrestricted use distribution and reproduction in any medium, provide the original author and source are credited.
INTRODUCTION
Dengue virus (DENV) is a major threat to worldwide human health. Over 50 million cases are reported per year, it is most prevalent in tropical and sub-tropical countries, itconsists of four serotypes, DENV1 – DENV4. DENV is a member of the Flaviviridae family (Chen et al.,2013; Dwivedi et al.,2017.). The transmission of the virus to humans is by a vector that isamosquito, primary vector Aedesaegypti,and secondary vector Aedesalbopictus. The viral genomes consist of a positive single-stranded RNA encoding for ten proteins; three structural proteins capsid (C), membrane protein (M), envelope protein (E), and seven non-structural proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. These proteins are translated as a polyprotein, which is cleaved into individual proteins during maturation by the host proteases, ( Behura and Severson, 2013, Sim et al.,2015, Sim and Hibberd, 2016 Pollett et al.,2018).
Chikungunya virus (CHIKV) is a member of the Togaviridaefamilygenus alphavirusit is a single stranded, enveloped, positive-sense RNA virus (Galán et al.,2015). The genome is 12 kb in size and contains two open reading frames encoding non-structural and structural proteins CHIKV has caused several outbreaks in several Southeast Asian countries and emerge as a severepublic health concern (Dutta et.al.,2018). It is also anarthropod-borne virus, the mode of transmission is the mosquitoes of the Aedes spp. where Aedesaegypti and Aedesalbopuctus serve as primary and secondary vectors and humans serve as hosts similar to the DENV virus (Volk et al.,2010, Wimalasiri et al.,2019).
All amino acids, except methionine and tryptophan, are coded by more than one codon. This phenomenon of using more than one codon for each amino acid is termed as synonymous codon usage (Jun-Jun et al.,2013). The synonymous codons do not occur equally they follow a pattern in which some codons are preferred over others and this is termed as codon usage bias (Zhou et al.,2012; Hemert and Berkhout,2016). Several of factors are known to add to codon usage bias, such as translational and transcriptional selection, mutational bias, protein structure, RNA stability, GC content, tRNA abundance, etc.
The study of codon usage bias can provide insights into the molecular evolution of these viruses (Butt et al., 2014).There are individual studies on the codon usage bias of DENV and CHIKV but here, we have performed a comparative study between the codon usage bias of these two important viruses with their common vector and host. Considering the increase in the occurrence of DENV and CHIKV comparative analysis of codon usage bias will be important for understanding viral evolution, as there is no specific drug and effective vaccine for DENV and CHIKV it is very important to analyse and understand the molecular evolution of the virus (Wimalasiri et al.,2019). .
The literature studies reflect that much research work has been done for the codon usage analysis of CHIKV and DENV independently, though the CHIKV and DENV are members of two different families but share common host and vector. And this important correlation between the two pathogen is almost not considered. There is a lacuna towards the understanding between the two pathogen and their vector and host.Very few studies are available which address the three components of transmission cycle of virus i.e. virus, vector and host in the study, but again not for the pathogens from different origin but sharing common host and vector.
An attempt has been made to understand the relationship between the codon usage of virus vector and host. So far, no work has been done to understand and compare the codon usage pattern of viruses from two different families. Therefore, understanding the role of codon usage in viruses, host and vector interaction will explore the relationship of different virus with common vector and host. This relationship will give a detailed insight towards understanding the evolutionary pattern and relationship between different genotypes of virus. An attempt has been taken to include the three components of transmission cycle of virus that are virus, vector and host in the study.
MATERIAL AND METHODS
Sequence data: Sequences: Complete genome sequences of Dengue (DENV-1 90, DENV-2 94 and DENV-3 105, DENV-4 93) and CHIKV (641) viruses were downloaded from the National Centre for Biotechnology (NCBI) database (http: //www.ncbi.nlm. nih.gov) in FASTA format. The accession numbers of the selected genomes were listed in the table (Supplementary Table.1).
Compositional Analysis: To understand the frequencies of occurrence of each nucleotide composition analysis was conducted. The overall frequency of occurrence of the nucleotides (A %, C %, U %, and G %) was calculated along with the frequency of each nucleotide at the third site of the synonymous codons (A3, C3, U3, and G3). The termination codon UAA, UAG, and UGA do not encode any amino acids and codons AUG and UGG are the only codons for Met and Trp, respectively, therefore, these five codons are excluded from the analysis(Lobo et al.,2009; Feng et al.,2013; Hemert and Berkhout 2016).
Relative synonymous codon usage (RSCU):The RSCU values of codons for DENV and CHIKV were calculated to determine the characteristics of synonymous codon usage. The synonymous codons with RSCU values > 1.0 have positive codon usage bias, while those with RSCU values < 1.0 have negative codon usage bias. When the RSCU value is 1.0, it means there is no codon usage bias for that amino acid and the codons are chosen equally. The synonymous codons with RSCU values >1.6 and <0.6 were treated as over-represented and under-represented codons, respectively. The RSCU values for codon were calculated using the formula given below: ![]() Where gij is the observed number of the ith codon for the jth amino acid, which has ni types of synonymous codons ( Zhong et al.,2007; Mune et al.,2017).
Where gij is the observed number of the ith codon for the jth amino acid, which has ni types of synonymous codons ( Zhong et al.,2007; Mune et al.,2017).
RESULTS AND DISCUSSION
Nucleotide composition of 382 sequences of DENV and 641 sequences of CHIKV was analyzed to understand its effect on preference for one type of codon over another, can be influenced greatly by the overall nucleotide composition of genomes (Jenkins et.al.,2003) and the nucleotide composition study reveals that codon usage pattern is influenced greatly by the overall nucleotide composition of genomes (Jun-Jun et al.,2013; Butt,et al.,2014).So First, the nucleotide composition of four serotypes of the DENV genome followed by the CHIKV genome was analyzed. Shown in (table 1)
We have found that the pattern of nucleotide composition of these two viruses is quite similar. In (table1) the mean nucleotide of A% of all the 4 serotypes of DENV and CHIKV was highest followed by G%, the mean nucleotide of C% is lowest in DENV and with the U% is lowest in CHIKV. The mean composition of GC and GC3 of all 4 serotypes of DENV and CHIKV virus were also given in (Table 1). The study of codon usage patterns of viruses can disclose more useful information about overall viral survival and evolution (Singh etal.,2016).In this study the pattern of nucleotide composition of these two viruses is quite analogous. This research seems to propose that there might be equal distribution of A,U,G and C nucleotides among codons of DENV and CHIKV , with a preference towards A-ended codon followed by G/C- ended codons (figure1).
Table 1.The mean of nucleotide composition analysis of DENV 1-4 and CHIKV genomes (%)
| Nucleotide composition (mean) | DENV1 | DENV2 | DENV3 | DENV4 | CHIKV |
| A% | 32.3 | 33.24 | 32.17 | 31.05 | 29.37 |
| G% | 25.85 | 25.22 | 25.92 | 26.28 | 25.37 |
| C% | 20.58 | 20.64 | 20.55 | 20.79 | 24.68 |
| U% | 21.54 | 21.08 | 21.35 | 21.89 | 20.44 |
| GC | 46.44 | 45.68 | 46.47 | 47.05 | 50.05 |
| GC3 | 45.73 | 46.64 | 43.55 | 48.13 | 48.70 |
Figure 1: Comparative Analysis Nucleotide Composition of all four serotypes of DENV and CHIKV Genomes: common pattern is observed in both the viruses the mean A% was the highest, followed by G%.
To understand the patterns of codon usage and evaluate the effect of nucleotide composition on codons usage pattern, we performed RSCU analysis and calculated the RSCU values (Table 2). Among the 16 most preferred codons in DENV genomes, six (UUG, CUG, GUG, GCC, AAC, and GAC) were G or C-ended (C-ended: 3; G-ended: 3) and the remaining ten (CUA, AUA, UCA, CCA, ACA, GCA, AAA, GAA, and AGA, GGA) were A-ended codons. There is no preferred codon ending with U. We observed that DENV exhibits higher codon usage bias towards G/C- and less towards A-ended codons.
Among 59 codons, six codons had RSCU value greater than or equal to 1.6 are over represented codons AGA (Arg), GGA (Gly), CCA (Pro), UCA (Ser), ACA (Thr), GUG (Val). And five under-represented codons had RSCU value equal to or less than 0.6 GCG (Ala), CGU (Arg), CCG (Pro), UCG (Ser), ACG (Thr). We observed that the codon usage pattern of virus and human host is quite similar in comparison to the codon usage pattern of virus and vector (Figure 2).
Among the 19 most preferred codons in CHIKV genomes, ten (UUC, CUG, AUC, GUG, GCC, UAC, UGC, CAC, AAC, and GAC) were G or C-ended (C-ended: 8; G-ended: 2) and the remaining seven (ACA, GCA, UCA, AGA, AAA, GAA, GGA and CCA, CAA) were A-ended codons (Table 2). None of the preferred codons were U-ended. We observed that CHIKV exhibits higher codon usage bias towards G/C- and less towards A-ended codons.
This indicates that the G/C content at the third position of the codons influences the codons usage patterns. Among 59 codons, only CUG (Leu) and AGA (Arg) had RSCU value greater than or equal to 1.6 are overrepresented codons. However, the underrepresented codons (RSCU,0.6), were identified as follows: UUA (Leu), GCG (Ala), and CGG (Arg). We observed that the codon usage pattern of virus and human host is quite similar in comparison to the codon usage pattern of virus and vector (Figure 2).
Figure 2: Comparative analysis of relative synonymous codon usage (RSCU) patterns. (1) Between DENV and CHIKV, Homo sapiens (HS) and Aedesaegypti (AG). (2) Between DENV, Homo sapiens (HS) and Aedesaegypti(AG) (3) Between CHIKV, Homo sapiens (HS) and Aedesaegypti (AG)
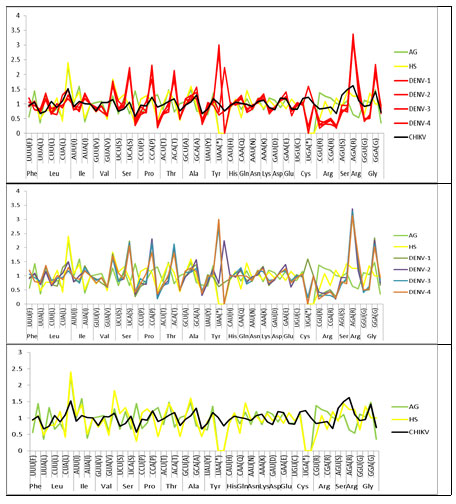
DENV and CHIKV both viruses belong to different families but share a common vector and host. When we analyzed the most abundantly used codons we observed a common pattern is both the viruses. The viruses exhibit a higher codon usage bias towards G/C- and less towards A-ended codons (Figure 2).
This suggests that GC composition plays an important role in crafting the codon usage pattern of both viruses. Another common feature we observed is that the codon usage pattern of virus and human host is quite similar in comparison to the codon usage pattern of virus and vector. The overall codon usage bias in DENV and CHIKV is low. The result is similar to other RNA viruses like polioviruses, H5N1 influenza virus, and SARS-Covs, respectively, we assume that the weak codon bias in RNA virus is advantageous to replicate efficiently in host cells (Hu et.al 2011; Huipeng et .al, 2019).
In this study, the comparative analysis of RSCU of DENV and its vector and host, CHIKV and its vector and host, and DENV and CHIKV was done to understand the patterns of codon usage and evaluate the effect of nucleotide composition on the codons usage pattern. Both the viruses exhibit a higher codon usage bias towards G/C- and less towards A-ended codons. This suggests that GC composition plays an important role in crafting the codon usage pattern of both viruses. The findings propose that even though RNA viruses have a high mutation rate, DENV and CHIKV has evolved with a moderately stable genetic composition at specific levels of codon usage.
The jist of, combining nucleotide composition and RSCU analysis, it is determined that the selection for preferred codons is influenced by nucleotide compositional constraints, which also suggests the presence of mutational pressure. However, it is not the claimed that the nucleotide compositional constraints are the sole factor associated with codon usage patterns in DENV and CHIKV, because though the RSCU values could disclose the codon usage pattern genome of DENV and CHIKV, (Hu et.al 2011; Huipeng et .al, 2019).
Table 2. The synonymous codon usage patterns of DENV and CHIKV, its hosts (Human) and vector (Aedes aegypti).
| AA | Codon | AG | HS | DENV-1 | DENV-2 | DENV-3 | DENV-4 | CHIKV |
| Phe | UUU(F) | 0.56 | 0.92 | 0.95 | 0.9 | 1.06 | 1.2 | 0.94 |
| UUC(F) | 1.44 | 1.08 | 1.05 | 1.1 | 0.94 | 0.8 | 1.06 | |
| Leu | UUA(L) | 0.36 | 0.48 | 0.71 | 0.69 | 0.81 | 0.76 | 0.67 |
| UUG(L) | 1.32 | 0.78 | 1.17 | 1.15 | 1.31 | 1.37 | 0.75 | |
| CUU(L) | 0.66 | 0.78 | 0.69 | 0.65 | 0.84 | 0.7 | 1.08 | |
| CUC(L) | 0.84 | 1.2 | 0.64 | 0.91 | 0.91 | 1.01 | 0.88 | |
| CUA(L) | 0.54 | 0.42 | 1.3 | 1.1 | 0.95 | 0.87 | 1.1 | |
| CUG(L) | 2.28 | 2.4 | 1.5 | 1.5 | 1.18 | 1.3 | 1.52 | |
| Ile | AUU(I) | 0.99 | 1.08 | 0.82 | 0.79 | 0.96 | 0.94 | 0.9 |
| AUC(I) | 1.59 | 1.41 | 0.82 | 1.01 | 0.74 | 0.88 | 1.08 | |
| AUA(I) | 0.39 | 0.51 | 1.36 | 1.19 | 1.3 | 1.17 | 1.01 | |
| AUG(M) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Val | GUU(V) | 1.04 | 0.72 | 0.89 | 0.85 | 0.84 | 0.74 | 0.77 |
| GUC(V) | 1.08 | 0.96 | 0.74 | 0.92 | 0.93 | 0.93 | 1.05 | |
| GUA(V) | 0.6 | 0.48 | 0.6 | 0.59 | 0.59 | 0.62 | 1.04 | |
| GUG(V) | 1.28 | 1.84 | 1.78 | 1.64 | 1.64 | 1.7 | 1.15 | |
| Ser | UCU(S) | 0.66 | 1.14 | 0.84 | 0.82 | 0.8 | 1.1 | 0.75 |
| UCC(S) | 1.2 | 1.32 | 1.09 | 0.89 | 0.98 | 0.89 | 0.83 | |
| UCA(S) | 0.66 | 0.9 | 2.23 | 2.04 | 2.17 | 2.07 | 1.06 | |
| UCG(S) | 1.44 | 0.3 | 0.26 | 0.35 | 0.44 | 0.42 | 0.57 | |
| Pro | CCU(P) | 0.68 | 1.16 | 0.63 | 0.73 | 0.9 | 0.72 | 0.96 |
| CCC(P) | 0.84 | 1.28 | 0.74 | 0.7 | 0.75 | 1.08 | 0.93 | |
| CCA(P) | 1.2 | 1.12 | 2.29 | 2.31 | 2.17 | 1.84 | 1.22 | |
| CCG(P) | 1.32 | 0.44 | 0.34 | 0.26 | 0.19 | 0.36 | 0.89 | |
| Thr | ACU(T) | 0.8 | 1 | 0.72 | 0.63 | 0.66 | 0.66 | 0.97 |
| ACC(T) | 1.48 | 1.44 | 0.96 | 0.84 | 0.73 | 1.04 | 1.09 | |
| ACA(T) | 0.72 | 1.12 | 1.82 | 2.03 | 2.13 | 1.82 | 1.17 | |
| ACG(T) | 1.01 | 0.44 | 0.51 | 0.5 | 0.48 | 0.48 | 0.77 | |
| Ala | GCU(A) | 1.08 | 1.08 | 0.96 | 0.96 | 1.13 | 1.17 | 0.96 |
| GCC(A) | 1.48 | 1.6 | 1.27 | 1.16 | 1.13 | 1.29 | 1.07 | |
| GCA(A) | 0.76 | 0.92 | 1.39 | 1.57 | 1.27 | 1.16 | 1.3 | |
| GCG(A) | 0.68 | 0.44 | 0.38 | 0.31 | 0.47 | 0.38 | 0.67 | |
| Tyr | UAU(Y) | 0.64 | 0.88 | 1.04 | 0.77 | 0.89 | 0.97 | 0.83 |
| UAC(Y) | 1.36 | 1.12 | 0.96 | 1.23 | 1.11 | 1.03 | 1.17 | |
| UAA(*) | 0 | 0 | 0.62 | 0.73 | 2.75 | 3 | 1.01 | |
| UAG(*) | 0 | 0 | 0.77 | 2.24 | 0.03 | 0 | 0.76 | |
| His | CAU(H) | 0.84 | 0.84 | 0.89 | 1.04 | 0.91 | 1 | 0.94 |
| CAC(H) | 1.16 | 1.16 | 1.11 | 0.96 | 1.09 | 1 | 1.06 | |
| Gln | CAA(Q) | 0.82 | 0.54 | 1.16 | 1.23 | 1.29 | 1.01 | 1.02 |
| CAG(Q) | 1.18 | 1.46 | 0.84 | 0.77 | 0.71 | 0.99 | 0.98 | |
| Asn | AAU(N) | 0.8 | 0.94 | 0.89 | 0.97 | 0.83 | 0.83 | 0.92 |
| AAC(N) | 1.2 | 1.06 | 1.11 | 1.03 | 1.17 | 1.17 | 1.08 | |
| Lys | AAA(K) | 0.8 | 0.86 | 1.34 | 1.3 | 1.16 | 1.24 | 1.12 |
| AAG(K) | 1.2 | 1.14 | 0.66 | 0.7 | 0.84 | 0.76 | 0.88 | |
| Asp | GAU(D) | 1.12 | 0.92 | 0.89 | 0.85 | 0.87 | 0.89 | 0.8 |
| GAC(D) | 0.88 | 1.08 | 1.11 | 1.15 | 1.13 | 1.11 | 1.2 | |
| Glu | GAA(E) | 1.16 | 0.84 | 1.21 | 1.4 | 1.17 | 1.25 | 1.17 |
| GAG(E) | 0.84 | 1.16 | 0.79 | 0.6 | 0.83 | 0.75 | 0.83 | |
| Cys | UGU(C) | 0.84 | 0.92 | 1.04 | 0.99 | 1 | 0.94 | 0.81 |
| UGC(C) | 1.16 | 1.08 | 0.96 | 1.01 | 1 | 1.06 | 1.19 | |
| UGA(*) | 0 | 0 | 1.6 | 0.03 | 0.23 | 0 | 1.23 | |
| UGG(W) | 0 | 0 | 1 | 1 | 1 | 1 | 1 | |
| Arg | CGU(R) | 1.38 | 0.48 | 0.3 | 0.42 | 0.28 | 0.18 | 0.83 |
| CGC(R) | 1.26 | 1.08 | 0.41 | 0.37 | 0.32 | 0.33 | 0.86 | |
| CGA(R) | 1.2 | 0.66 | 0.51 | 0.45 | 0.31 | 0.44 | 0.89 | |
| CGG(R) | 1.02 | 1.2 | 0.26 | 0.18 | 0.21 | 0.21 | 0.68 | |
| Ser | AGU(S) | 0.96 | 0.9 | 0.74 | 0.95 | 0.7 | 0.8 | 1.3 |
| AGC(S) | 1.11 | 1.44 | 0.83 | 0.94 | 0.91 | 0.72 | 1.5 | |
| Arg | AGA(R) | 0.64 | 1.26 | 3.15 | 3.37 | 3.3 | 3.14 | 1.62 |
| AGG(R) | 0.54 | 1.26 | 1.36 | 1.2 | 1.58 | 1.71 | 1.11 | |
| Gly | GGU(G) | 1.12 | 0.64 | 0.48 | 0.46 | 0.41 | 0.5 | 0.92 |
| GGC(G) | 1.04 | 1.36 | 0.51 | 0.55 | 0.63 | 0.54 | 0.95 | |
| GGA(G) | 1.48 | 1 | 2.34 | 2.26 | 2.12 | 2.03 | 1.42 | |
| GGG(G) | 0.36 | 1 | 0.67 | 0.74 | 0.84 | 0.93 | 0.71 |
CONCLUSION
The study shows that the comparative analysis of the codon usage pattern in DENV and CHIKV is slightly biased. The nucleotide composition and mutation pressure act as a key factor in shaping codon usage patterns in both DENV and CHIKV. Our data suggested that the codon usage pattern of DENV and CHIKV is evolving to re-adapt to different vector and hosts. Though, further detailed analysis is required to understand the relationship of codon choices between viruses and hosts. To the best of our knowledge, this is the first study comparing the codon usage of DENV and CHIKV with their common host and vector and it is anticipated to increase our understanding of the evolution of DENV and CHIKV.
However, The information from this research may not only help to understand the molecular evolution of these two viruses, but it will also contribute to the further development of the virus vaccine and antiviral drug (from Genome to Hit).To achieve the goal for viruses ; the preferred codon information is going to play a key regulating role to develop a therapeutics. It is well stated that the preferred codon are conserved throughout the species, whereas the remaining genome content of virus is highly unstable that is susceptible to the environment.
Ethical Statement: This article does not contain any studies with human participants or that were performed on animals.
REFERENCES
Behura S. K. and Severson D. W (2013) Nucleotide substitutions in dengue virus serotypes from Asian and American countries insights into intra codon recombination and purifying selection BMC microbiology vol 13 p 37
Butt A. M, Nasrullah I and Tong Y (2014) Genome-wide analysis of codon usage and influencing factors in Chikungunya viruses PloS one vol 9 p 90905
Chen H.T, Gu Y.X and Y.S.Liu (2013) Analysis of synonymous codon usage in Dengue Viruses J Anim Vet Adv vol. 12 p 88-98
Dwivedi V.D, Tripathi I.P, Tripathi R.C, Bharadwaj S. and Mishra S.K (2017)Genomics proteomics and evolution of dengue virus briefings in Functional Genomics vol 16 p 217-227
Dutta S.K, Bhattacharya T and Tripathi A (2018) Chikungunya virus genomic microevolution in Eastern India and its in-silico epitope prediction 3 Biotech Vol 8 p 318
Feng C, Xu C.j, Wang Y, Liu W.l, Yin X.R, Li X, et al (2013) Codon usage patterns in Chinese bayberry (Myrica rubra) based on RNA-Seq data BMC genomics vol 14 p732
Galán-Huerta K. A, Rivas-Estilla A. M, Fernández-Salas I, Farfan-Ale J. A, and Ramos-Jiménez J (2015) Chikungunya virus A general overview medicina universitaria vol 17 p 175-183
Hemert F. Van and Berkhout B (2016) Nucleotide composition of the Zika virus RNA genome and its codon usage virology journal vol 13 p 95
Huipeng Yao, Mengyu Chen, and Zizhong Tang (2019) Analysis of Synonymous Codon Usage Bias in Flaviviridae Virus vol 2019
Jenkins GM, Holmes EC (2003) The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res 92: p 1–7.
Hu, JS Q.-Q.Wang, J. Zhang et al (2011) The characteristic of codon usage pattern and its evolution of hepatitis C virus, Infection, Genetics and Evolution, vol. 11, no. 8, p2098–2102,
Jun-Jun Ma F. Z, Zhang Jie, Zhou Jian Hua, Ma Li-Na, Ding Yao-Zhong, Chen Hao-Tia, Gu Yuan-Xing and Liu Yong-Sheng (2013) Analysis of Synonymous codon usage in Dengue viruses Journal of animal and veterinary advances vol. 12 p 88-98
Lobo F. P, Mota B. E, Pena S. D, Azevedo V, Macedo A. M, Tauch A et al( 2009) Virus-host coevolution common patterns of nucleotide motif usage in Flaviviridae and their hosts PloS one vol 4 p 6282
Mune A, Pandey A, and Pandey K. M, (2017) Genome-wide comparative analysis of the codon usage pattern in Flaviviridae family bioscience biotechnology research communication vol 10 p 680-688
Singh, NK A. Tyagi, R. Kaur, R. Verma, and P. K. Gupta,( 2016) Characterization of codon usage pattern and influencing factors in Japanese encephalitis virus Virus Research, vol. 221, p 58–65
Pollett S, Melendrez M. C, Berry I Maljkovic, Duchêne S, Salje H,Cummings D. A. T et al (2018) Understanding Dengue virus evolution to support epidemic surveillance and counter-measure development Infection Genetics and Evolution vol 62 p 279-295
Sim S, Aw P. P, A. Wilm, Teoh G, Hue K. D. T, Nguyen N. M et al ( 2015) Tracking Dengue virus intra-host genetic diversity during human-to-mosquito transmission PLoS neglected tropical diseases vol. 9 p 0004052
Sim S. and Hibberd M. L (2016) Genomic approaches for understanding Dengue insights from the virus vector and host genome biology vol 17 p 38-38
Volk S. M, Chen R, Tsetsarkin K. A, Adams A. P, Garcia T. I, Sall A. A (2010) et al Genome-Scale Phylogenetic Analyses of Chikungunya Virus Reveal independent emergences of recent epidemics and various evolutionary rates journal of virology vol 84 p 6497
Wimalasiri Yapa B. M. C. R, Stassen L, Huang X, Hafner L. M, Hu W, Devine G. J.et al (2019) Chikungunya virus in Asia Pacific a systematic review emerging microbes and infections vol 8 p 70-79
Xiang H, Zhang R, Butler III R. R, Liu T, Zhang L, Pombert J.F et al( 2015) Comparative analysis of codon usage bias patterns in microsporidian genomes PloS one vol 10 p 0129223
Zhou J.h, Gao Z. l, Zhang J, Chen H.t, Pejsak Z, Ma L.n, et al (2012) Comparative the codon usage between the three main viruses in Pestivirus genus and their natural susceptible livestock virus genes vol 44 p 475-481
Zhong J, Li Y, Zhao S, Liu S, and Zhang Z (2007) Mutation pressure shapes codon usage in the GC-Rich genome of foot-and-mouth disease virus virus genes vol 35 p 767-776

