
Biotechnological
Communication
Biosci. Biotech. Res. Comm. 10(4): 697-703 (2017)
16S metagenomic analysis and taxonomic distribution
of enriched microbial consortia capable of simultaneous
biodegradation of organochlorines by illumina platform
Saghee Madhu Raju
1,2
and Rajkumar Bidlan
2#
1
Research Scholar, Rayalaseema University, Kurnool, India
2
Dr. Bidlan’s Research Institute, Hyderabad, India
#
Present Address: Department of Biotechnology, Delhi Technological University, Shahbad Daulatpur, Delhi, India
ABSTRACT
Organochlorine pesticides are ubiquitous group of recalcitrant molecules that accumulate in food chains and have
inherent toxic effects and adverse health effects. To circumvent the problem, microbial communities are found to
be promising candidates for degrading the organochlorine pesticide’s and removal of residues. In this study, a novel
microbial consortium isolated from Yamuna and Godavari rivers capable of simultaneous biodegradation of organo-
chlorine pesticides (DDT and Lindane) was subjected to metagenomic sequencing. This consortia used was enriched
by progressively increasing concentrations of Lindane and DDT (organochlorine pesticides) for months till a stable
Lindane and DDT tolerant population was established, and found to be degrading mixture of organochlorine pes-
ticides with concentrations up to 30 ppm of DDT and Lindane. Currently, in the realm of our knowledge very few
metagenomic analysis were carried out to characterize the consortia and understand the biodiversity of microbial
communities in the riverine ecosystems, that was found to be unique and highly ef cient in bio-degradation of
organochlorine pesticides. The study concluded biodiversity with a shannon alpha-diversity index of 3.0317 and
identi ed 871 species with Brevundimonas diminuta (previously assigned to the genus Pseudomanas) having abun-
dance ratio of 17.57 % followed by Stenotrophomonas acidaminiphila in the mixed consortium and deciphered the
systematic and functional contexts within riverine metagenome.
KEY WORDS: MICROBIAL CONSORTIUM, BIOREMEDIATION, DICHLORODIPHENYLTRICHLOROETHANE, HEXACHLOROCYCLOHEXANE,
LINDANE, METAGENOMICS, AMPLICON, ILLUMINA
697
ARTICLE INFORMATION:
*Corresponding Author: madhu.saghee@gmail.com
Received 10
th
Oct, 2017
Accepted after revision 19
th
Dec, 2017
BBRC Print ISSN: 0974-6455
Online ISSN: 2321-4007 CODEN: USA BBRCBA
Thomson Reuters ISI ESC and Crossref Indexed Journal
NAAS Journal Score 2017: 4.31 Cosmos IF: 4.006
© A Society of Science and Nature Publication, 2017. All rights
reserved.
Online Contents Available at:
http//www.bbrc.in/
DOI: 10.21786/bbrc/10.4/13

698 16S METAGENOMIC ANALYSIS AND TAXONOMIC DISTRIBUTION OF ENRICHED MICROBIAL CONSORTIA BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS
Saghee Madhu Raju and Rajkumar Bidlan
INTRODUCTION
Organochlorine pesticides (OCPs) were excessively used
globally for pest control and agricultural purposes and
public health control (Aktar et al., 2009). OCPs are
ubiquitous group of recalcitrant molecules that degrade
slowly and accumulate through food chains (Amrita
et al., 2007) and produce a signi cant magni cation at
each tropic level. One of the major sinks for persistent
organic pollutants discharged into environment is the
water ecosystem i.e. rivers and lake beds. Organochlorine
pesticides were detected in rivers where higher concentra-
tions of Lindane, Endosulfan and DDT were found (Pandey
et al., 2011)and the residue presence was even detected
in drinking and bottled water (Mutiyar et al., 2011). It
is highly essential and vital to remove these pollutants
from the environment, from the sinks primarily water
and soil ecosystems to nally eliminate their residues.
Microorganisms are found to be potential degraders
of organochlorine compounds, notably water and soil
habitants belonging to genera Bacillus, Pseudomonas,
Arthrobacter, Klebsiella, Acinetobacter, Alcaligenes, Fla-
vobacterium and Micrococcus were found to be effective
bio-degraders (Ka lzadeh et al., 2014 Eric et al 2017).
In this paper, we present the ndings of metagenomic
analysis leveraging next-generation sequencing (NGS)
performed using Hiseq 2500 system (Kumar et al., 2015).
The Metagenomics was carried on the de ned microbial
consortium identi ed from water ecosystems, Yamuna
River (North India) and Godavari River (South India)
capable of simultaneous degradation of organochlorine
pesticides (Bidlan, 2003). The taxonomic distribution
and biodiversity among the microbial consortium was
established that comprised of interacting microbial pop-
ulations (Oulas A et al., 2015 Eric et al., 2017 ).
MATERIALS AND METHODS
Lindane -HCH (insecticidal isomer) was of 97% purity
and obtained from Sigma- Aldrich, USA. DDT, 99.4%
pure, was donated by Hindustan Insecticides Ltd, India.
All other chemicals and reagents used in the study were
of analytical grade and were purchased from standard
manufacturers. The microbial consortium subjected to
Metagenomic analysis was isolated from Yamuna (North
India) and Godavari rivers (South India) and enriched by
progressively increasing concentrations of Lindane and
DDT (organochlorine pesticides) for months till a stable
Lindane and DDT tolerant population was established in
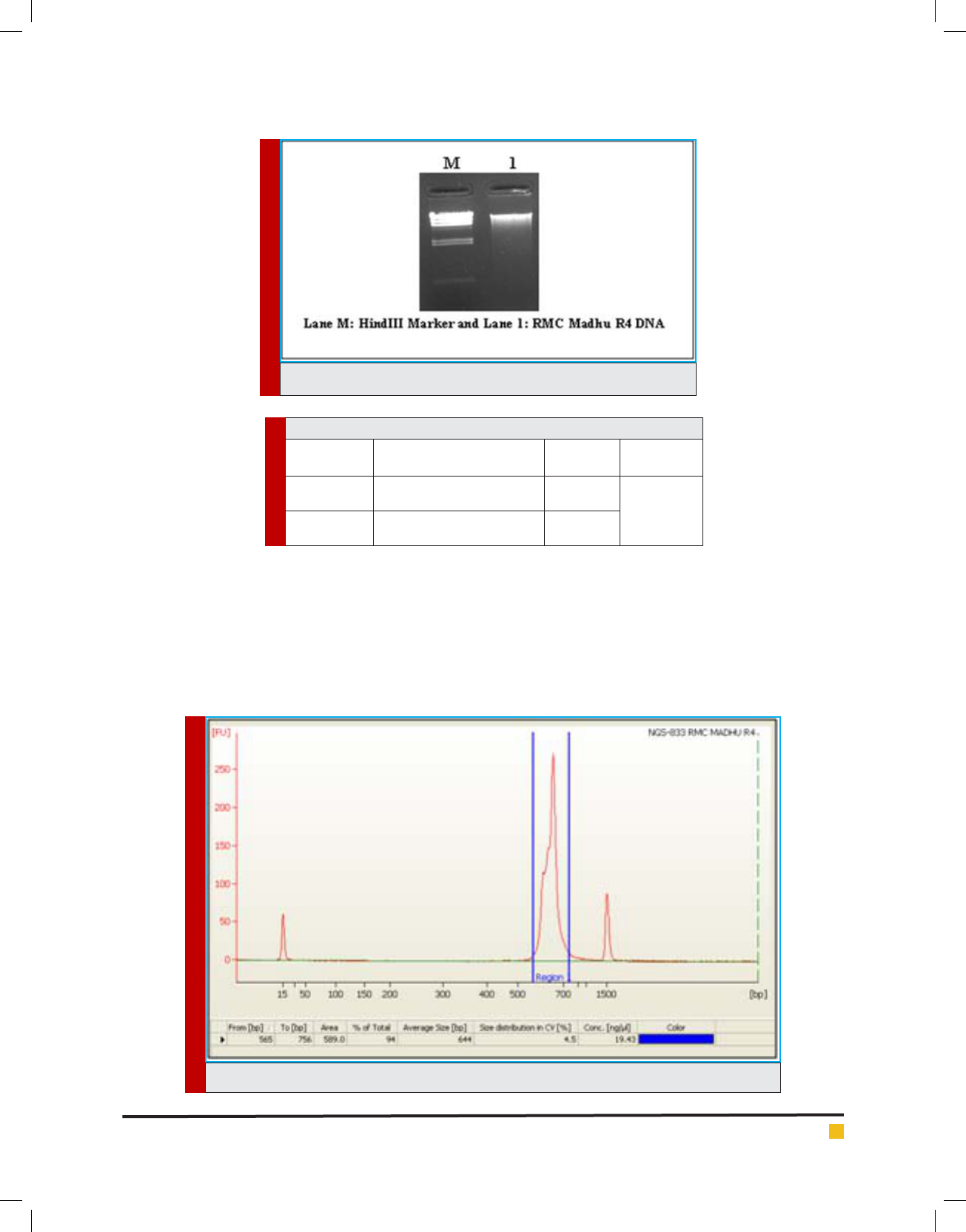
the ask (Bidlan 2003). DNA was isolated using Xcelgen
Bacterial gDNA kit and quality of gDNA was checked on
0.8 % agarose gel (loaded 5 l) for the single intact band.
The gel was run at 110 V for 30 min. 1 µl of each sample
was loaded in Nanodrop 8000 for determining A260/280
ratio. The DNA was quanti ed using QubitdsDNA HS
Assay kit (Life Tech). 1 µl of each sample was used for
determining concentration using Qubit® 2.0 Fluorometer
(Ogata et al., 1990).
The amplicon library was prepared using Nextera
XT Index Kit (Illuminainc) as per the 16S Metagenomic
Sequencing Library preparation (Eric J. et al., 2017).
Primers for the ampli cation of the V3-V4 hyper-var-
iable region of 16S rDNA gene of bacteria and Archaea
are used for this study (Table-1).
The amplicons with the Illumina adaptors were ampli-
ed by using i5 and i7 primers that add multiplexing
index sequences as well as common adapters required
for cluster generation (P5 and P7) as per the standard
Illumina requirements (Esling et al., 2015). The ampli-
con libraries were puri ed by 1X AMpureXP beads and
checked on Agilent High Sensitivity (HS) chip on Bio-
analyzer 2100 and quanti ed on uorometer by Qubi-
tdsDNA HS Assay kit (Life Technologies).
After obtaining the Qubit concentration for the library
and the mean peak size from Bioanalyser pro le, library
was loaded onto HiSeq 2500 at appropriate concentra-
tion (10-20 pM) for cluster generation and sequencing
(Sharpton, 2014). Paired-End sequencing allows the tem-
plate fragments to be sequenced in both the forward and
reverse directions. Kit reagents were used in binding of
samples to complementary adapter oligos on ow cell.
The adapters were designed to allow selective cleavage
of the forward strands after re-synthesis of the reverse
strand during sequencing. The copied reverse strand was
then used to sequence from the opposite end of the frag-
ments (Blomquistet al., 2013).
The libraries were prepared from sample after ampli-
fying the V3-V4 region of the 16S segment. Size of
library was 644 bp and the library was sequenced using
the Illumina sequencing chemistry to generate ~150
Mb of data per sample. The next generation sequenc-
ing (NGS) for the sample was performed on the Illumina
platform, HiSeq 2500 (Kumar et al., 2015).
Paired end sequence stitching was carried out for
sample using FLASH (Fast Length Adjustment of Short
reads) with parameter minimum overlap of 10 bases to
merge paired-end reads from next-generation sequenc-
ing experiments (Tanja et al., 2011). QIIME (Quantitative
Insight into Microbial Ecology) was used for analyzing
16S metagenome data from NGS platforms and, is imple-
mented in python language (Kuczynski et al., 2011). Chi-
meras composed of DNA from two or more microbial
species which are artifacts made during the PCR process.
They were ltered rst, using usearch61 algorithm (de
novo, abundance-based), from the Flashed/stitched data
then taken for analysis. A total of 2,44,283 non chi-
meric sequences from sample were used for OTU pick.
In the next step, the similar sequences were clustered,
BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS 16S METAGENOMIC ANALYSIS AND TAXONOMIC DISTRIBUTION OF ENRICHED MICROBIAL CONSORTIA 699
Saghee Madhu Raju and Rajkumar Bidlan
FIGURE 1. QC of gDNA on 0.8% agarose gel
Table 1. Primers used in the Study
Oligo Name Oligo Sequence (5’ to 3’)
Length of
primer
Product size
(Approx.)
Prokaryote
V3-Forward
CCTACGGGNBGCASCAG
17
~ 460 bps
Prokaryote
V4-Reverse
GACTACNVGGGTATCTAATCC
21
FIGURE 2. Bioanalyzer pro le of the Consortium on DNA 1000 chip.
i.e., sequences coming from the same genus, together
into one representative taxonomic unit called as Opera-
tional Taxonomic Unit (OTU). The basis of this sequence
clustering is 97% sequence similarity and implemented
through UCLUST algorithm. OTU-picking identi ed
highly similar sequences across the samples and pro-
vided a platform for comparisons of community struc-
ture. All the sequences from all the samples were clus-
tered into Operational Taxonomic Units (OTUs) based on
their sequence similarity.
A representative sequence was selected for each of
these OTU’s picked. As these OTU’s made up of a group of
sequences, they were represented through one sequence
to assign a taxonomic name to the group. Thus repre-

Saghee Madhu Raju and Rajkumar Bidlan
700 16S METAGENOMIC ANALYSIS AND TAXONOMIC DISTRIBUTION OF ENRICHED MICROBIAL CONSORTIA BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS
FIGURE 3. Taxonomic distribution of de ned consortium at genus level showing the
relative abundance of each genus within microbial community.
Table 2. Relative Abundance of the
genus in the consortium
Taxonomy (Genus) Abundance
Brevundimonas 17.60%
Enterococcus 8.50%
Leucobacter 3.90%
Lysinibacillus 2.90%
Alcaligenes 1.40%
Table 3. Alpha-diversity
Sample shannon
Observed
species
chao1
Consortiaenriched with
pesticides
3.0317 871 871
FIGURE 4. Rank abundance plot of Microbial Consortium
sentative set of OTUs were prepared which consist of
2,911 sequences. With representative sequence in hand,
the taxonomic names to these sequences were assigned
at 90% sequence similarity. This is done using UCLUST
algorithm, where query is representative sequences
and subjects that are curated sequences at greengenes
database.
RESULTS AND DISCUSSION
Diversity calculation for each sample was performed
and compared the types of communities, using the taxo-
nomic assignments.
-DIVERSITY
-Diversity or within-sample diversity is calculated
using an OTU table which gives idea about species
richness. Alpha diversity summarizes the diversity of
organisms in a sample using different metrics in a habi-
Saghee Madhu Raju and Rajkumar Bidlan
BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS 16S METAGENOMIC ANALYSIS AND TAXONOMIC DISTRIBUTION OF ENRICHED MICROBIAL CONSORTIA 701
FIGURE 5. An OTU table heat map showing taxonomy assignment. The OTU heatmap
displays raw OTU counts per sample, where the counts are colored based on the contri-
bution of each OTU to the total OTU count present in that sample (blue: contributes low
percentage of OTUs to sample; red: contributes high percentage of OTUs).
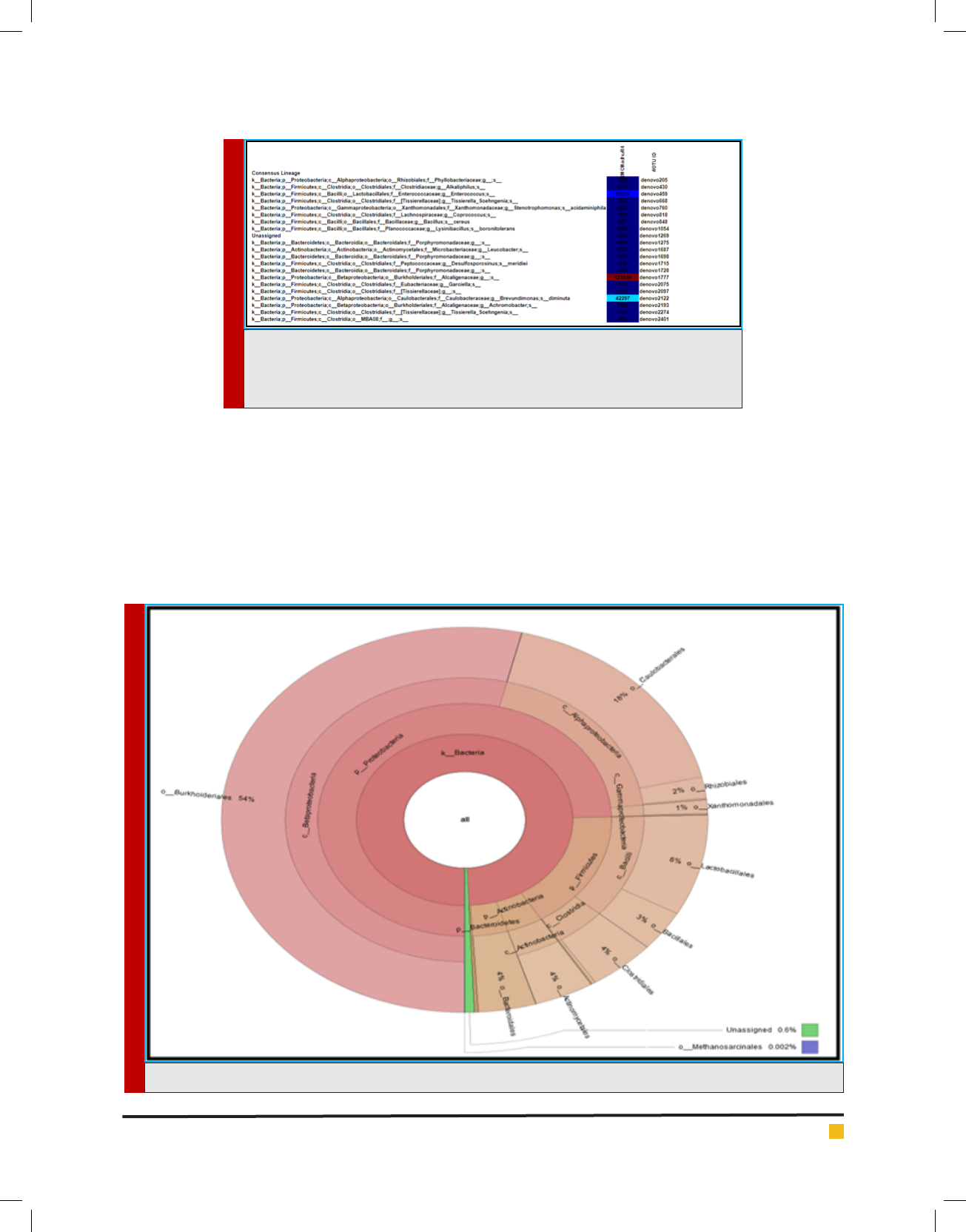
FIGURE 6. Krona graph for taxonomy assignment for Microbial Consortium at order level.
tat/sample. The below table summarizes the -Diversity,
where the columns correspond to alpha diversity met-
rics and the rows correspond to samples and their cal-
culated diversity measurements (Lozupone, Catherine
et al., 2007).
The rank abundance curve representing species rich-
ness and species evenness is shown in Figure 4. Species
richness can be viewed as the number of different spe-
cies on the chart and species evenness is derived from
the slope of the line that ts the graph.
The OTU table was developed to visualise as a
heatmap where each row corresponds to an OTU and
each column corresponds to a sample. The higher the
relative abundance of an OTU in a sample, the more
intense the color at the corresponding position in the
heatmap.

Saghee Madhu Raju and Rajkumar Bidlan
702 16S METAGENOMIC ANALYSIS AND TAXONOMIC DISTRIBUTION OF ENRICHED MICROBIAL CONSORTIA BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS
Krona graph tool was used to display abundance and
hierarchy simultaneously using a radial space- lling
display. The Krona chart features a red-green colour
gradient signifying average value within each taxon
(Ondov et al., 2011)
CONCLUSION
The metagenomic sequencing comprehensively sam-
pled all genes in all organisms present in microbial
consortia and evaluated bacterial diversity and abun-
dance of microbes (Table-4). This study also identi ed
at genotypic level any unculturable microorganisms
that are otherwise dif cult or impossible to analyze
(Handelsman J. et al., 2004). The study concluded bio-
diversity with a shannon alpha-diversity of 3.0317 and
identi ed 871 species genotypically, with Brevundi-
monas diminuta having abundance ratio of 17.57 %
followed by Stenotrophomonas acidaminiphila in the
mixed consortium. This consortia characterized was
found to be degrading mixture of organochlorine pes-
ticides with concentrations up to 30 ppm of DDT and
Lindane con rmed by GC-MS/MS. Although research
has been carried out using on single strain and sin-
gle compound of organochlorines, the current study
data provides an insight on how bacterial communities
in mixed consortia are taxonomically distributed and
their biodiversity. The metagenomic characterization
identi ed the consortia in a de nitive manner which
acts as promising solution for bioremediation of organ-
ochlorine mixtures.
NCBI Sequence Accession Number: DNA sequences
obtained have been deposited at National Center for Bio-
technology Information (NCBI) Sequence Read Archive
under the bioproject ID PRJNA420925 and accession
codeSRX348847.
ACKNOWLEDGEMENTS
The authors would like to acknowledge the support of
Xcelris Labs for extending support, wherever required
during the research and Rayalaseema University and UGC
for support and encouragement for the research studies.
REFERENCES
Akhtar MW, Dwaipayan Sengupta, and Ashim Chowdhury,
2009 Impact of pesticides use in agriculture: their bene ts and
hazards, Interdisciplinary Toxicology, 2(1) pp.1–12.
Amrita Malik, Kunwar P. Singh, Priyanka Ojha, 2007 Residues
of Organochlorine Pesticides in Fish from theGomti River,
India, Bulletin of Environmental Contamination and Toxicol-
ogy, Volume 78, Number 5, Page 335
Bidlan R. 2003 Studies on DDT degradation by Bacterial strains.
In: Isolation, puri cation and identi cation of microbes capa-
ble of DDT- degradation. Ph.D. thesis, University of Mysore,
India.pp 90-142. 2003.
Bidlan R. and Manonmani H.K., 2004 Aerobic degradation of
dichlorodiphenyltrichloroethane (DDT) by Serratiamarcescens
DT-1P.Process Biochemistry,38, pp.49-56.
BlomquistTM, CrawfordEL, LovettJL, YeoJ, StanoszekLM.
(2013) Correction: Targeted RNA Sequencing with Competi-
tive Multiplex-PCR Amplicon Libraries. PLOS ONE 8(12): 10.
1371
Eric J. de Muinck,Pål Trosvik, Gregor D. Gil llan,
JohannesR.Hov and ArvindY.M.Sundaram 2017 A novel
ultra-high-throughput 16S rRNA gene amplicon sequenc-
ing library preparation method for the Illumina HiSeq plat-
form, Microbiome 5:68 https://doi.org/10.1186/s40168-017-
0279-1
Garza DR, Dutilh BE 2015 From cultured to uncultured genome
sequences: metagenomics and modeling microbial ecosys-
tems. Cellular and Molecular Life Sciences. 72:4287-4308.
doi:10.1007/s00018-015-2004-1.
Handelsman J. 2004 Metagenomics: Application of Genomics
to Uncultured Microorganisms. Microbiology and Molecular
Biology Reviews. 68(4):669-685. doi:10.1128/MMBR.68.4.669-
685.2004.
Ka lzadeh F, Ebrahimnezhad M, Tahery Y. 2015 Isolation and
Identi cation of Endosulfan-Degrading Bacteria and Evalua-
tion of Their Bioremediation in Kor River, Iran.Osong Public
Health and Research Perspectives. 6(1):39-46. doi:10.1016/j.
phrp.2014.12.003.
Kuczynski J, Stombaugh J, Walters WA, González A, Caporaso
JG, Knight R. 2011 Using QIIME to analyze 16S rRNA gene
sequences from Microbial Communities.Current protocols
in bioinformatics / editoral board, Andreas D Baxevanis
CHAPTER:Unit10.7. doi:10.1002/0471250953.bi1007s36.
Table 4. Organisms Identi ed through
Metagenomic Characterization by Hiseq2500,
Illumina Platform, NGS
Total Reads 5,88,408
Total number of stitched reads 2,81,957
Number of OTUs 2,911
Abundant phylum Proteobacteria
Abundant class Betaproteobacteria
Abundant order Burkholderiales
Abundant family Alcaligenaceae
Abundant genus Brevundimonas
Abundant species dimunita
shannon alpha-diversity 3.0317
Observed species 871

Saghee Madhu Raju and Rajkumar Bidlan
BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS 16S METAGENOMIC ANALYSIS AND TAXONOMIC DISTRIBUTION OF ENRICHED MICROBIAL CONSORTIA 703
Kumar S, Krishnani KK, Bhushan B, Brahmane MP. 2015
Metagenomics: Retrospect and Prospects in High Through-
put Age. Biotechnology Research International. 2015:121735.
doi:10.1155/2015/121735.
Lozupone, Catherine et al. 2007 Quantitative and qualitative
beta diversity measures lead to different insights into factors
that structure microbial communities. Applied and environ-
mental microbiology 73 5 1576-85.
Mutiyar, PK A. K. Mittal and A. Pekdeger 2011 Status of organ-
ochlorine pesticides in the drinking water well- eld located in
the Delhi region of the ood plains of river Yamuna, Drink.
Water Eng. Sci., 4, pp.51–60.
Ogata, M., Mattern, R., Schneider, P.M. et al. Z Rechtsmed
(1990) 103: 397. https://doi.org/10.1007/BF01263148
Ondov, B.D., Bergman, N.H. & Phillippy, A.M. 2011 BMC Bioin-
formatics 12: 385. https://doi.org/10.1186/1471-2105-12-385
Oulas A, Pavloudi C, Polymenakou P. 2015 Metagenomics:
Tools and Insights for Analyzing Next-Generation Sequenc-
ing Data Derived from Biodiversity Studies. Bioinformatics and
Biology Insights. 9:75-88. doi:10.4137/BBI.S12462.
Pandey, P P. S. Khillare, Krishan Kumar 2011 Assessment of
Organochlorine Pesticide Residues in the Surface Sediments of
River Yamuna in Delhi, India, Journal of Environmental Pro-
tection 2, 511-524
Philippe Esling, Franck Lejzerowicz, Jan Pawlowski 2015 Accu-
rate multiplexing and ltering for high-throughput amplicon-
sequencing, Nucleic Acids Research, Volume 43, Issue 5, 11
March 2015, Pages 2513–2524, https://doi.org/10.1093/nar/
gkv107
Sharpton TJ. 2014 An introduction to the analysis of shot-
gun metagenomic data.Frontiers in Plant Science. 5:209.
doi:10.3389/fpls.2014.00209.
Tanja Mago
c
ˇ
, Steven L. Salzberg 2011 FLASH: fast length
adjustment of short reads to improve genome assemblies,
Bioinformatics, Volume 27, Issue 21, 1 November 2011, Pages
2957–2963, https://doi.org/10.1093/bioinformatics/btr507