
Virological
Communication
Biosci. Biotech. Res. Comm. 11(2): 300-306 (2018)
A comparative analysis of overall codon usage pattern
of Louping Ill virus with natural livestock host and
associated vector
Anjusha Mune
1
*, Ajay Pandey
2
and Khushhali Menaria Pandey
1
*
1
Department of Biological Science and Engineering, MANIT Bhopal (M.P.) India
2
Department of Applied Mechanics, MANIT Bhopal (M.P.) India
ABSTRACT
Louping ill is a zoonotic viral disease caused by louping ill virus (LIV) which is a member of genus Flavivirus in the
family Flaviviridae. This febrile illness to livestock can further develop into fatal encephalitis .The virus LIV is closely
related to tick-borne encephalitis virus and occurs wherever the primary vector tick (Ixodes ricinus) is found. To
understand the viral evolution, comparison and analysis of the codon usage of LIV, its vector, and the host is impor-
tant. The present study reports the pattern of codon usage in LIV, its vector, and the host by calculating the Effective
number of Codons (ENC), Codon Adaptation Index (CAI), and Relative Synonymous Codon Usage (RSCU) and other
indicators. The results indicate relatively low codon usage bias of LIV. The ENC - plot demonstrates the substantial
role played by mutation pressure. The com parative analysis of CAI among virus, vector and its host, indicates that
the virus is more adaptive to the host than the vector. A comparative analysis of RSCU between virus, vector, and its
host shows that the codon usage pattern of LIV is a mix of coincidence and antagonism. To the best of our knowledge,
this is the rst report describing codon usage analysis of LIV and ndings are expected to increase our understanding
of factors involved in viral evolution and tness toward vector and host.
KEY WORDS: CODON USAGE, EVOLUTION, LOUPING ILL VIRUS (LIV), EFFECTIVE NUMBER OF CODONS, RELATIVE SYNONYMOUS CODON
USAGE
300
ARTICLE INFORMATION:
*Corresponding Author: anjumune@gmail.com
Received 10
th
March, 2018
Accepted after revision 19
th
May, 2018
BBRC Print ISSN: 0974-6455
Online ISSN: 2321-4007 CODEN: USA BBRCBA
Thomson Reuters ISI ESC / Clarivate Analytics USA and
Crossref Indexed Journal
NAAS Journal Score 2018: 4.31 SJIF 2017: 4.196
© A Society of Science and Nature Publication, Bhopal India
2018. All rights reserved.
Online Contents Available at: http//www.bbrc.in/
DOI: 10.21786/bbrc/11.1/16

Anjusha Mune et al.
INTRODUCTION
Louping ill virus (LIV) is a tick-born member of the genus
Flavivirus in Flaviviridae family. It is a positive single
stranded, 40-50 nm RNA virus whose genome comprises
a single open reading frame (ORF) that is approximately
11 kb in length (Grard et al.,2007;Jeffries et al., 2014).
The ORF encodes a polyprotein that consists of three
structural and seven non-structural proteins. The virus
show high degree of genetic homology to tick-borne
encephalitis virus (TBEV) of the same family (McGuire
et al., 1998; Jiang et al., 1993). It is mainly transmitted
by ticks and the primary vector is Ixodes ricinus (Dobler
et al., 2010).LIV mainly causes febrile illness in sheep,
cattle, horse, pigs and some other animals that may
eventually result in fatal encephalitis.
Sheep are the most important reservoir host for LIV.
The disease is dominantly detected in animals from
upland areas of British Isles (Gao et al., 1997) though
the disease is also reported in Scotland, Ireland, and
northern England where the tick vector Ixodes ricinus
is found. Infection with LIV was rst reported in sheep
of Basque region of northern Spain in 1987 (Gonzalez
et al., 1987). Most of the cases of LI infection occur
in spring / early summer when ticks are common. In
endemic areas morbidity and mortality depends upon
animal’s immune status, concurrent infection and other
factors. All age group of animal get infected by it and
once encephalitis is developed the case fatality rate goes
up to 50%. The mortality rate is even higher in animals
that are less than two years old. Currently, there is no
speci c treatment for LIV with only supportive therapies
being helpful to some extent (Hyde et al., 2007 Mans-
eld et al., 2015 Butt et al., 2016).
The molecular sequence data started to be accu-
mulated nearly 20 years ago. It was observed that the
genetic code is redundant and most amino acids can be
translated by more than one codon (Wang et al., 2011).
This redundancy is a key factor regulating the ef ciency
and accuracy of protein production.Alternative codons
within the same group that encode the same amino acid
are often called ‘synonymous’ codons. These codons are
not randomly selected within and between genomes.
This is referred to as ‘codon usage bias’ (CUB). CUB are
widespread across the tree of life and are in uenced by
mutation pressure, natural or translational selection,
secondary protein structure, replication, selective tran-
scription, hydrophobicity and hydrophilicity of the pro-
tein, and the external environment (Xiang et al., 2015
Butt et al., 2016 Mune et al., 2017).
As viruses are intracellular pathogens they have to
co-evolve with host molecular mechanisms. The inter-
play between the codon usage of the virus and its host is
expected to affect the overall viral survival, tness, evasion
of the host immune system and evolution. The knowledge
of the codon usage of viruses can provide information
about their molecular evolution and extend our under-
standing of the regulation of viral gene expression. This
may also offer signi cant improvement in vaccine design
for which the ef cient expression of viral proteins may be
required to generate immunity (Tao et al., 2009 Velazquez
et al., 2016). To gain insight into the characteristics of the
viral genome and evolution, the codon usage patterns of
the three components of transmission cycle, namely - the
virus (LIV), vector (Ixodes ricinus), and hosts (Sheep (Ovis
aries), Pig (Sus scrofa) and cattle (Bos taurus)) were inves-
tigated in our study.
MATERIALS AND METHODS
SEQUENCE DATA
The complete genome sequences were downloaded from
the National Centre for Biotechnology (NCBI) data-
base (http: //www.ncbi.nlm. nih.gov) in FASTA format.
The detailed information (accession numbers, coun-
try, sequence length etc.) of the selected genomes were
listed [Table. S1]. Open reading frames (ORF) of all the
genomic sequences were identi ed by using NCBI ORF
nder (https://www.ncbi.nlm.nih.gov/orf nder/). The
host (Ovis aries, Sus scrofa and Bos taurus) and vec-
tor (Ixodes ricinus) codon usage were obtained from the
Codon Usage Data Base (CUD).
CODON USAGE ANALYSIS
The overall frequency of occurrence of the nucleotides
(A %, C %, U %, and G %) was calculated along with
the frequency of each nucleotide at the third site of the
synonymous codons (A
3
, C
3
, U
3
and G
3
).Also the overall
GC, AU and GC
3
content were calculated using MEGA7
software to investigate the compositional properties of
coding region of LIV. To investigate the codon usage
pattern, the RSCU (Relative synonymous codon usage)
values for synonymous codons were calculated accord-
ing to the published equation (Sharp et al., 1986). The
stop codons (UAA, UAG and UGA) and AUG for Met,
UCG for Try were not introduced into the RSCU anal-
ysis. Further, ENC (Effective number of codon) values
were calculated to measure the magnitude of codon
usage bias in the coding sequences of viral genome. The
ENC value ranges from 20 (when only one synonymous
codon is chosen by the corresponding amino acid) to 61
(when all synonymous codons are used equally). A low
ENC value indicates a strong codon usage bias (Wright
et al., 1990; Zhang et al., 2011 Butt et al., 2013).
The CAI (Codon adaptation index) was used to esti-
mate the adaptation of LIV to its host and vector codons.
BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS A COMPARATIVE ANALYSIS OF OVERALL CODON USAGE PATTERN OF LOUPING ILL VIRUS WITH 301
Anjusha Mune et al.
CAI values range from 0 to 1. A higher CAI score for a
given gene indicates more similarity between its codon
usage and the prede ned reference set, using the CAIcal
approach (available at: http://genomes.urv.es/CAIcal)
(Puigbo et al., 2008).
RESULTS AND DISCUSSION
SYNONYMOUS CODON USAGE IN LIV
The preference for one type of codon over another can
be greatly in uenced by the nucleotide composition of
genome. We rst analysed nucleotide composition and
observed that the nucleotides A and G were higher and
followed by C and U (Table 1) The LIV genome is rich
with G content having a mean value of 32.17. For a bet-
ter understanding we analysed nucleotide composition
at third position of codon and observed the dominance
of G
3
nucleotide with a mean value of 34.20. Even the
percentage of dinucleotide with G is higher compared
to dinucleotide with other nucleotides (respective mean
values for GC, AU, GC
3
and AU
3
being 54.74, 45.26,
60.74, and 39.26).
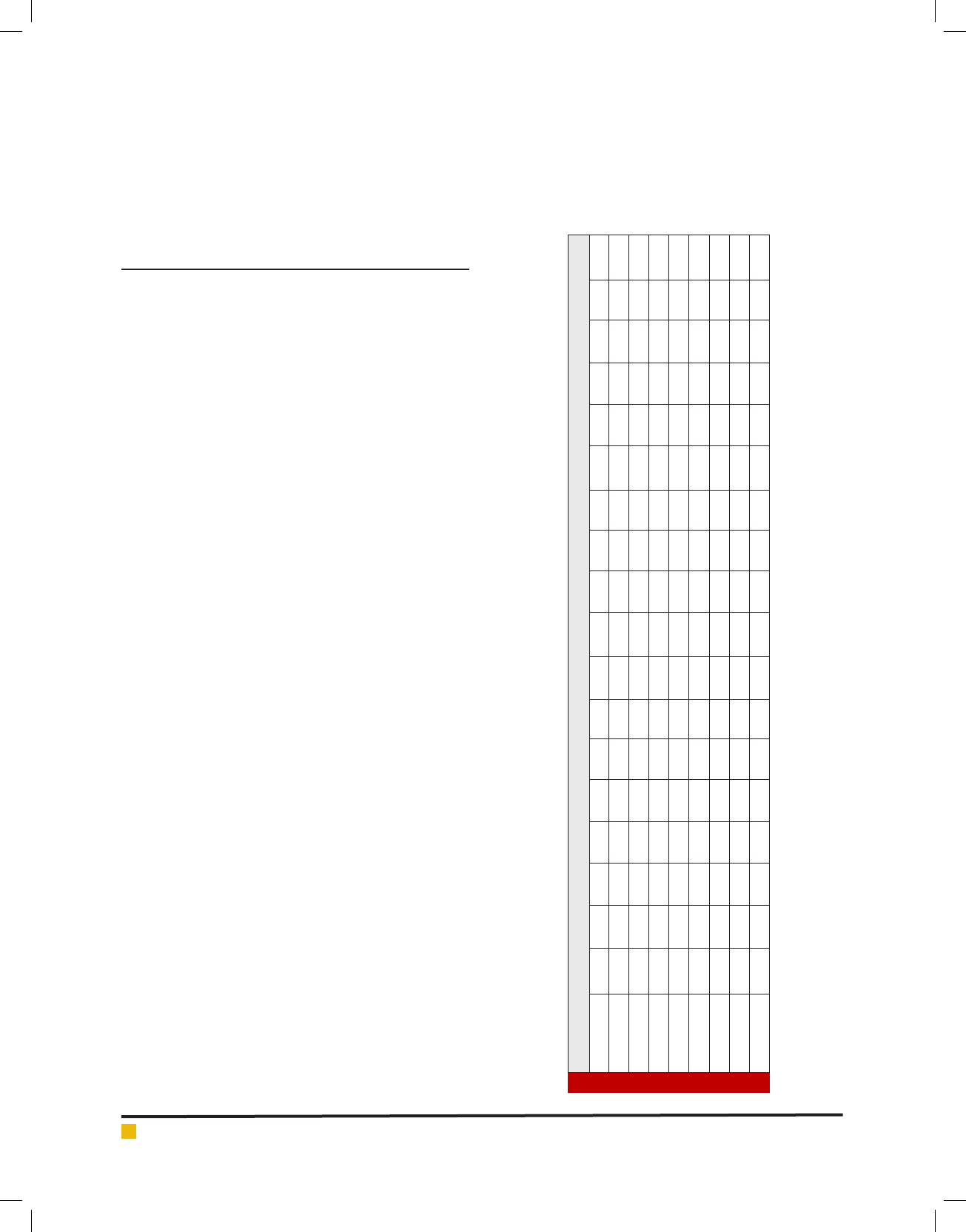
To investigate the extent of codon usage bias, the
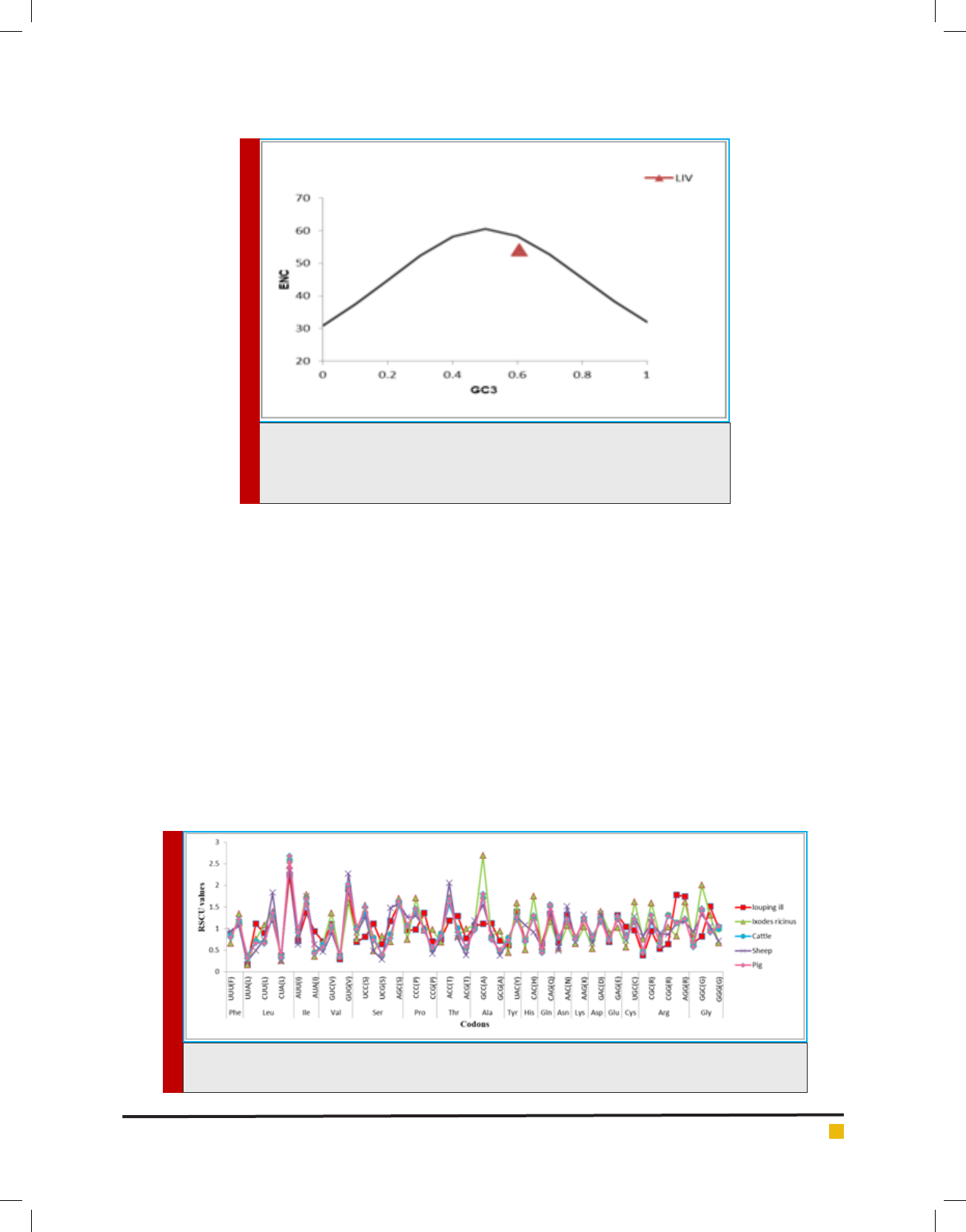
ENC values among LIV genome were calculated. An
average value of 53.97 represents stable ENC value
(ENC > 40) (Mune et al., 2017) which suggests that the
genomic composition of LIV is conserved. The result
shows that the codon usage of LIV is slightly biased and
mainly affected by the nucleotide composition. To fur-
ther understand the codon usage pattern, the analysis of
ENC - plot (ENC value V/s GC
3
content) was carried out.
It is observed that all points lie below the expected curve
(Fig.1). This implies that the codon usage bias is mainly
affected by nucleotide composition (in other words - by
mutation pressure).
To further explore the codon usage preferential opti-
mization and adaptation of LIV in relation to its vector
and hosts CAI analysis was performed. CAI values were
calculating keeping Ixodes ricinus, Ovis aries,Sus scrofa
and Bos taurus codon usage as a reference set. A mean
CAI value of 0.658 was obtained for the LIV ORFs in
relation to primary vector Ixodes ricinus codon usage
reference set and mean CAI values of 0.623, 0.689 and
0.711 were obtained for the LIV ORFs in relation to host
pig , sheep and cattle (Ovis aries,Sus scrofa and Bos tau-
rus) codon usage reference set respectively. In this study
we found a tendency for higher CAI values indicating
lower ef ciency of translation. A comparison between
vector and host indicated a lower CAI for LIV in relation
to pig, which leads to lower ef ciency of protein syn-
thesis in pig. This suggests that the interplay of codon
usage between LIV and its hosts may in uence viral t-
ness, survival and evolution.
Table 1. Nucleotide composition analysis of LIV genome (IR: Ixodes ricinus, SS: Sus scrofa, OA: Ovis aries, BT: Bos taurus)
Accession no. U C A G U3 C3 A3 G3 AU GC AU3 GC3 GC12 ENC CAI
IR
CAI
SS
CAI
OA
CAI
BT
NC_001809.1 20.72 22.67 24.47 32.13 18.57 26.53 20.85 34.06 45.19 54.81 39.41 60.59 30.29 53.88 0.658 0.622 0.691 0.711
Y07863 20.72 22.67 24.47 32.13 18.57 26.53 20.85 34.06 45.19 54.81 39.41 60.59 30.29 53.88 0.658 0.622 0.691 0.711
KT224354.1 20.81 22.48 24.57 32.14 18.62 26.47 20.67 34.23 45.38 54.62 39.30 60.70 30.35 54.12 0.657 0.623 0.687 0.711
KP144331.1 20.61 22.59 24.60 32.20 18.16 26.68 20.94 34.23 45.21 54.79 39.09 60.91 30.45 53.89 0.658 0.624 0.689 0.711
KJ495985 20.81 22.48 24.58 32.13 18.62 26.47 20.67 34.23 45.39 54.61 39.30 60.70 30.35 54.15 0.657 0.623 0.687 0.710
KF056331.1 20.74 22.54 24.45 32.27 18.48 26.56 20.59 34.38 45.19 54.81 39.06 60.94 30.47 53.93 0.658 0.622 0.690 0.711
Avg. 20.74 22.57 24.52 32.17 18.50 26.54 20.76 34.20 45.26 54.74 39.26 60.74 30.37 53.97 0.658 0.623 0.689 0.711
Std. D 0.0722 .0890 .0652 .0561 .1780 .0756 .1361 .1234 .0961 .0961 .1532 .1532 .0766 .1261 .0005 .0008 .0018 .0004
302 A COMPARATIVE ANALYSIS OF OVERALL CODON USAGE PATTERN OF LOUPING ILL VIRUS WITH BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS
Anjusha Mune et al.
FIGURE 2. Comparative analysis of relative synonymous codon usage (RSCU) patterns between virus, vector
and three hosts (cattle, sheep and pig).
FIGURE 1. Graph showing the relationship between the effective number of codons
(ENC) and GC content of the third codon position (GC3).The curve indicates the
expected codon usage if GC compositional constraints alone account for codon
usage bias.
To investigate the codon usage pattern of virus, an RSCU
analysis was performed for the 59 sense codons (Table.2). In
LIV among the 18 most abundantly used codons, 12 were
G/C-ended ( ve G-ended, seven C-ended) and the remain-
ing six were A/U-ended ( ve A-ended and one U-ended).
To determine the potential in uences of the vector
and host on the codon usage pattern of the LIV, the
RSCU pattern of LIV coding sequence were correlated
with those of Ixodes ricinus (vector) and pig, sheep and
cattle (hosts) (Fig.2).All the 18 most abundantly used
codons of vector and host were G/C ending (In Ixodes
ricinus twelve C-ended and six G-ended, Pig thirteen
C-ended and ve G-ended, cattle twelve C-ended and
six G-ended, and in sheep eleven C-ended codons six
G-ended codons and one U-ended codon) we observed
a common pattern of preference towards G/C-ended
codons in vector and host. An analysis of over and
under - represented codons showed that for LIV 4 out of
18 preferred codons (CUG for Leu, GUG for Val and AGA
and GGA for Arg) in Ixodes ricinus 11 out of 18 preferred
codons (CUG for Leu, AUC for Ile, GUG for Val, AGC for
Ser, CCC for Pro, ACC for Thr, GCC for Ala, CAC for His,
UGC for Cys, AGG for Arg and GGC for Gly), in cattle 3
out18 preferred codons (CUG for Leu, GUG for Val and
GCC for Ala), in sheep 5 out of 18 preferred codons (CUG
and CUC for Leu, AUC for Ile, GUG for Val and ACC for
Thr), and in pig 6 out of 18 preferred codons (CUG for
Leu, AUC for Ile, GUG for Val, AGC for Ser and ACC for
Thr, GCC for Gly) had RSCU value >1.6, whereas the
remaining preferred codons had RSCU values >0.6 and
<1.6. CUG for Leu and GUG for Val are common over-
represented codons in virus vector and hosts.
BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS A COMPARATIVE ANALYSIS OF OVERALL CODON USAGE PATTERN OF LOUPING ILL VIRUS WITH 303

Anjusha Mune et al.
Table 2. The relative synonymous codon usage patterns of LIV, its host (cattle,
sheep and pig) and primary transmission vector (Ixodes ricinus)
AA Codon Pathogen Vector Host
louping ill Ixodes ricinus Cattle Sheep Pig
Phe UUU 0.88 0.66 0.85 0.94 0.79
UUC 1.12 1.34 1.15 1.06 1.21
Leu UUA 0.18 0.16 0.38 0.24 0.32
UUG 1.11 0.75 0.71 0.49 0.67
CUU 0.90 1.08 0.7 0.74 0.65
CUC 1.2 1.40 1.26 1.83 1.35
CUA 0.37 0.26 0.36 0.24 0.33
CUG 2.24 2.45 2.59 2.46 2.68
Ile AUU 0.71 0.85 0.98 0.63 0.91
AUC 1.36 1.79 1.57 1.74 1.67
AUA 0.93 0.36 0.45 0.63 0.42
Val GUU 0.7 0.68 0.64 0.46 0.57
GUC 1.1 1.36 1.01 0.91 1.07
GUA 0.29 0.35 0.4 0.36 0.34
GUG 1.92 1.61 1.95 2.27 2.03
Ser UCU 0.69 0.76 1.04 0.91 0.99
UCC 0.81 1.54 1.37 1.28 1.5
UCA 1.11 0.48 0.79 0.48 0.73
UCG 0.64 0.83 0.39 0.28 0.39
AGU 1.17 0.69 0.87 1.48 0.77
AGC 1.58 1.70 1.53 1.58 1.62
Pro CCU 0.96 0.75 1.08 1.26 1.05
CCC 0.98 1.70 1.39 1.29 1.46
CCA 1.36 0.96 1 1.03 0.94
CCG 0.7 0.98 0.53 0.42 0.56
Thr ACU 0.75 0.68 0.89 0.78 0.83
ACC 1.18 1.71 1.55 2.05 1.68
ACA 1.29 0.82 1.01 0.78 0.92
ACG 0.77 1.00 0.56 0.38 0.57
Ala GCU 1.06 1.07 1 1.18 0.96
GCC 1.11 2.69 1.71 1.55 1.8
GCA 1.12 0.84 0.8 0.9 0.74
GCG 0.72 0.95 0.48 0.37 0.5
Tyr UAU 0.61 0.45 0.79 0.72 0.73
UAC 1.39 1.59 1.21 1.28 1.27
His CAU 0.75 0.50 0.75 1.08 0.7
CAC 1.25 1.75 1.25 0.92 1.3
304 A COMPARATIVE ANALYSIS OF OVERALL CODON USAGE PATTERN OF LOUPING ILL VIRUS WITH BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS

Anjusha Mune et al.
Gln CAA 0.66 0.60 0.46 0.57 0.44
CAG 1.34 1.16 1.54 1.43 1.56
Asn AAU 0.68 0.55 0.81 0.49 0.79
AAC 1.32 1.07 1.19 1.51 1.21
Lys AAA 0.79 0.65 0.78 0.68 0.76
AAG 1.21 1.04 1.22 1.32 1.24
Asp GAU 0.8 0.54 0.84 0.66 0.8
GAC 1.2 1.40 1.16 1.34 1.2
Glu GAA 0.69 0.91 0.78 0.75 0.72
GAG 1.31 1.02 1.22 1.25 1.28
Cys UGU 1.04 0.57 0.85 0.72 0.79
UGC 0.96 1.62 1.15 1.28 1.21
Arg CGU 0.38 0.75 0.49 0.82 0.44
CGC 0.94 1.59 1.17 1.15 1.31
CGA 0.53 0.80 0.68 0.89 0.6
CGG 0.64 1.04 1.32 0.86 1.29
AGA 1.78 0.83 1.14 1.12 1.12
AGG 1.74 1.62 1.2 1.16 1.23
Gly GGU 0.66 0.78 0.64 0.92 0.57
GGC 0.82 2.01 1.43 1.33 1.46
GGA 1.51 1.31 0.95 1.05 0.91
GGG 1.02 0.67 0.99 0.71 1.05
Supplementary Table 1. Detail information about the LIV
Strain
Name
Virus
Type
GenBank
Accession
Sequence
Length
ORF ORF
Length
Collection
Date
Host GenBank
Host
Country
369/T2 LIV NC_001809 10871 130-10374 10245 -N/A- Unknown -N/A- -N/A-
369/T2 LIV Y07863 10871 130-10374 10245 -N/A- Unknown -N/A- -N/A-
LEIV-7435Tur LIV KT224354 10829 106-10350 10245 -N/A- Tick Hyalomma
marginatum
(tick)
Turkmenistan
LI3/1 LIV KP144331 10880 133-10377 10245 1962 Sheep Ovis aries United
Kingdom
Primorye-
185-91
LIV KJ495985 10871 129-10373 10245 07/22/1991 Human Homo
sapiens
Russia
Penrith LIV KF056331 10875 132-10376 10245 2009 Sheep Ovis aries United
Kingdom
None of the preferred codons were under-represented
(RSCU<0.6). UUA and CUA for Leu and GUA for Val
are common underrepresented codons in virus, vector
and hosts. Interestingly, a mixture of coincidence and
antagonism was observed in the codon usage pattern
as LIV showed no complete coincidence or complete
antagonism to any of the patterns of its vector and host.
Among the 18 most abundantly used codons, the ratio of
coincident/antagonist preferred codon was 12:6 between
virus vector and hosts.
CONCLUSION
Our analysis has provided an insight into codon usage
pattern of LIV virus and its relationship with host and
vector. We observed that the codon usage bias of LIV is
BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS A COMPARATIVE ANALYSIS OF OVERALL CODON USAGE PATTERN OF LOUPING ILL VIRUS WITH 305

Anjusha Mune et al.
slightly biased which re ects that the key role played by
mutation pressure and natural selection. Our observa-
tions suggest that codon usage of LIV is an evolutionary
process However, a more comprehensive analysis with
higher sample sizes is needed as this study and sub-
sequent analysis is based on a relatively small sample
size.
REFERENCES
Butt AM, Nasrullah I, Qamar R, Tong Y. (2016) Evolution of
codon usage in Zika virus genomes is host and vector speci c.
Emerg Microbes Infect. 5(10):e107.
Butt A.M., Nasrullah I., Tong Y. (2013) Genome-Wide Analy-
sis of Codon Usage and In uencing Factors in Chikungunya
Viruses. PLoS One 9(3): e90905, Vol.9
Dobler G. (2010) Zoonotic tick-borne aviviruses, Vet Micro-
biol. vol. 140(3-4):221-8.
Gao GF, Zanotto PM, Holmes EC, Reid HW, Gould EA. (1997)
Molecular variation, evolution and geographical distribution
of louping ill virus. ActaVirol.41 (5):259-68.
Gonzalez L, Reid HW, Pow I, Gilmour JS. (1987) A disease
resembling louping-ill in sheep in the Basque region of Spain.
Vet Rec. 121(1), 12-3.
Grard G., Moureau G., Charrel RN, Lemasson JJ., et al.(2007)
Genetic characterization of tick-borne aviviruses: new
insights into evolution, pathogenetic determinants and tax-
onomy. Virology.
Jeffries CL., Mans eld KL., Phipps LP., Wakeley PR., et al.
(2014) Louping ill virus: an endemic tick-borne disease of
Great Britain, J Gen Virol.vol 95, 1005-1014.
Jiang WR, Lowe A, Higgs S, Reid H, Gould EA. (1993) Sin-
gle amino acid codon changes detected in louping ill virus
antibody-resistant mutants with reduced neuro virulence., J
Gen Virol.74, 931-5.
Mans eld KL, Morales AB, Johnson N, AyllónN., et al. (2015)
Identi cation and characterization of a novel tick-borne Fla-
vivirus subtype in goats (Capra hircus) in Spain. J Gen Virol.
vol. 96, 1676-1681.
McGuire K., Holmes EC., Gao GF., Reid HW., Gould EA. (1998)
Tracing the origins of louping ill virus by molecular phyloge-
netic analysis. J Gen Virol , vol-79 pg 981-988.
Mune A., Pandey.A., Pandey.K.M.(2017) Genome-wide com-
parative analysis of the codon usage pattern in Flaviviridae
family, Biosci. Biotech. Res. Comm. 10(4): 680-688
Puigbò P, Bravo IG, Garcia-Vallvé S. (2008) E-CAI: a novel
server to estimate an expected value of Codon Adaptation
Index (eCAI). BMC Bioinformatics.29; 9:65.
Sharp PM, Tuohy TM, Mosurski KR. (1986) Codon usage in
yeast: cluster analysis clearly differentiates highly and lowly
expressed genes. Nucleic Acids Res. 14(13):5125-43.
Tao P, Dai L, Luo M, Tang F, Tien P, Pan Z.(2009) Analysis of
synonymous codon usage in classical swine fever virus. Virus
Genes, 38, 104-12.
Velazquez-Salinas L, Zarate S, Eschbaumer M, Pereira Lobo F,
et.al .(2016) Selective Factors Associated with the Evolution of
Codon Usage in Natural Populations of Arboviruses. PLoS One,
11(7):e0159943.
Wang M, Liu YS, Zhou JH, Chen HT, YX, Zhang J., et al. (2011)
Analysis of codon usage in Newcastle disease virus. Virus
Genes, Vol 42, 245-53.
Wright F. (1990) the ‘effective number of codons’ used in a
gene, Gene, Vol.87, 23-29.
Xiang H, Zhang R, Butler RR 3rd, Liu T, Zhang L, Pombert JF,
Zhou Z. (2015) Comparative Analysis of Codon Usage Bias Pat-
terns in Microsporidian Genomes. PLoS One. 9;10(6):e0129223.
Zhang J, Wang M, Liu WQ, Zhou JH, Chen HT, Ma LN, Ding
YZ, Gu YX, Liu YS.(2011) Analysis of codon usage and nucleo-
tide composition bias in polioviruses. Virol J., 8:146.
306 A COMPARATIVE ANALYSIS OF OVERALL CODON USAGE PATTERN OF LOUPING ILL VIRUS WITH BIOSCIENCE BIOTECHNOLOGY RESEARCH COMMUNICATIONS